HealthManagement, Volume 23 - Issue 2, 2023
 PRINT OPTIMISED
PRINT OPTIMISED
Effective regulatory systems are an essential component of health system strengthening and contribute to better public health outcomes, even more so in the age of medical devices that are enabled by machine learning.
Key Points
- Without medical devices, many critical healthcare procedures would not be possible.
- WHO provides guidance to its Member States on how to regulate medical devices to ensure access to latest technologies, while minimizing risk of harm to patients and device users.
- Collectively, WHO and other regulators are seeking to ensure that machine learning-enabled medical devices are monitored quality, safety and performance throughout the life cycle.
Introduction
Medical devices are essential in carrying out various healthcare procedures, including diagnosis, prevention, monitoring treatments, supporting people living with disabilities, and intervening in and treating of acute and chronic illnesses.
Medical devices are used in diverse settings, from personal use at home, to advanced medical facilities, or remote (small) clinics where healthcare professionals and clinicians’ practise. Without medical devices, many critical healthcare procedures would not be possible.
Currently, there are an estimated two million different kinds of medical devices on the world market, categorised into more than 22 000 generic devices groups. Such diversity underscores the need for freely accessible global nomenclature for medical devices (WHO Executive Board 2019).
Effective regulatory systems are an essential component of health system management and contribute to better public health outcomes.
While national authorities have been regulating medicines for the past 70 years, the regulation of medical devices began 30 years later.
The objective of regulating medical devices is to ensure that patients have access to safe and effective medical devices of high quality, while also preventing products that offer limited clinical benefits or pose a safety risk from entering the market. Medical device regulation requires stakeholders (manufacturers, importers and distributors) to meet the requirements set down by regulatory authorities based on internationally recognised standards.
However, with the increasing digitalisation of healthcare through machine-learning-based medical devices (ML/MDs), traditional models of change assessment and post-market surveillance present some new challenges. Despite these challenges, the principles of medical device regulation still apply to ML/MDs when they have a medical purpose (IMDRF 2023).
Many low- or lower-middle-income countries lack the resources, awareness, and commitment required to successfully transition from an unregulated market to a basic medical device regulatory framework.
To this end, WHO implements various strategies to facilitate national and regional efforts to regulate medical devices.
WHO Support for Regulation of Medical Devices
Effective regulatory oversight is a critical requirement for ensuring that products in the market continue to be of good quality, safety, and appropriate performance throughout the lifecycle from pre-market, placing on the market, to post-market and disposal or decommissioning.
However, regulatory authorities can be constrained in terms of resources, awareness, commitment and priority setting, which means legal provisions, guidelines, and/or procedures for medical devices can be inadequate. WHO supports regulatory authorities and manufacturers to ensure quality, safety and performance of medical devices through many activities.
WHO’s work on regulation and safety priorities are based on a strategic plan entitled “Delivering quality-assured medical products for all 2019-2023: WHO’s five-year plan to help build effective and efficient regulatory systems” (WHO 2019).
The WHO Global Model Regulatory Framework for Medical Devices including in vitro diagnostic medical devices (GMRF) recommends guiding principles and uniform definitions and specifies the elements of effective and efficient regulations to be embodied within binding/enforceable national laws.
Many countries have neither the financial resources nor the technical expertise to move from a minimally regulated market directly to one with a comprehensive medical devices law and regulatory controls. The GMRF recommends instead a stepwise approach to regulating the quality, safety and performance of medical devices. This staged development starts from basic-level regulatory controls – such as the publication of the law, import controls, and resourcing the regulatory authority to take enforcement actions – then progresses to expanded-level regulatory controls – such as inspection of registered establishments and oversight of clinical investigations.
Not all countries will be able to move at the same pace or devote the same levels of resources, systematic assessment and continued progress in this area. Over time, however, this will lead to greater public confidence in the regulation, as well as safety, performance and quality of medical devices including in vitro diagnostics (IVDs) used in health systems.
WHO Good Regulatory Practices help regulators to execute core regulatory functions, by leveraging the competencies of policymakers, procurers, distributors, clinicians, patients and consumers. WHO Good Reliance Practices also describe the various models of work-sharing, recognition, and reliance that may be leveraged (WHO 2021).
To evaluate the strengths and weaknesses of their regulatory systems, national authorities can use WHO’s Global Benchmarking Tool, recently adapted for medical devices as the GBT + medical devices (WHO). The evaluation findings from GBT can be used to create plans for institutional development of regulatory system.
To assist countries adopt reliance principles for regulation, WHO Prequalification (PQ) can provide relevant information on products. While not used for regulatory approval, PQ offers governments and international aid agencies valuable information for procuring medical products from an approved list that has been evaluated by subject matter experts.
Further information about WHO assessment of a particular device can be found in the WHO prequalification public reports (WHO).
WHO’s Support to Detect Substandard and Falsified Medical Devices
The vast majority of medical products globally circulate in markets with insufficient regulatory oversight to assure the quality of medical devices and prevent or detect the distribution of substandard/falsified medical devices.
WHO’s strategy to control substandard and falsified medical products involves a three-pronged approach: prevent, detect, and respond.
It is critical to empower users and patients to document the first sign that a device may be responsible for harm. Any health facility that uses medical devices should have a user feedback form available to all staff and a process to ensure the manufacturer (or their local authorized representative) is informed as soon as possible about a device failure or malfunctioning. Hospitals and other health facilities are likely to have their own systems for ensuring patient safety which can interface with medical device incident reporting for detection of substandard/falsified devices.
A crucial aspect of ensuring the quality, safety, and performance of medical devices is post-market surveillance (PMS). This is a set of activities conducted by manufacturers to continuously monitor the quality, safety and performance of medical devices throughout their lifecycle (ISO). Manufacturers gather and analyse data from user feedback (through complaints, technical support calls-outs, maintenance, installation, user training), scientific peer-reviewed scientific literature, and publicly available regulatory sources.
Users in health facilities, hospitals, diagnostic centers, and community clinics should report any device-related incident such as mortality or morbidity, involving the user or other person, or any deterioration or malfunction of a device to its manufacturer.
In turn, there are certain categories of incidents that must be reported by the manufacturer to the regulatory agencies, including incidents that caused harm or might have cause harm.
As part of the post-market surveillance analysis, manufacturers may need to undertake correction (fix the problem now), corrective action (prevent the same problem recurring) or preventive actions (prevent a problem from occurring in the first instance) to address reduce risks to patients, and users. From their perspective, regulatory authorities must take appropriate regulatory actions if manufacturers’ investigations or actions are inadequate or implement their own market surveillance activities, including trending of incidents. This includes overseeing any corrective actions taken by the manufacturer in the field, with the ultimate goal of safeguarding the interests of consumers and patients as part of the response to substandard or falsified devices.
WHO guidance and other internationally recognised standards encourage harmonised reporting to enhance data analysis for trends, and sharing of field safety notices in the public domain by regulators for users in healthcare facilities to freely access (WHO 2020; IMDRF please replace with:
WHO guidance and other internationally recognised standards encourage harmonised reporting to enhance data analysis for identifying trends. Additionally, they encourage the sharing of field safety notices by regulatory authorities in the public domain, ensuring that healthcare facilities and users have unrestricted access to this valuable information (WHO 2020; IMDRF 2020).
The WHO Global Surveillance and Monitoring System (GSMS) (WHO Global Surveillance and Monitoring System) works with regulatory authorities and other key stakeholders to improve the quantity, quality and analysis of data on SF medical products, and to use that data in the better prevention, detection and response to SF products, to protect public health. Its current scope includes medicines, vaccines and IVDs and certain medical devices listed by WHO. To date, all incidents for WHO prequalified IVDs are reported into GSMS, primarily by their manufacturers, any current field safety notices are displayed on the WHO’s website (WHO 2023).
WHO is also currently assessing the need for a freely accessible global database of field safety notices for all medical devices for their users.
Regulation of Machine-Learning Enabled Medical Devices
Machine learning (ML) is a subset of artificial intelligence (AI). AI uses algorithms or models to learn, make decisions and make predictions. Machine learning allows ML models to be developed by ML training algorithms through analysis of datasets to identify patterns without models being explicitly programmed to do so.
Medical devices can make use of ML to achieve their intended use.
If a ML product meets the definition of a medical device, it is considered to be a Machine Learning-enabled Medical Device, or ML/MD and would be regulated as such. Typically, algorithms tend to be “locked” and don’t change unless the regulators assess and clears any change. ML/MDs tend not to be “locked” and will continuously be updating the model. Therefore, traditional methods for change assessment of medical device in the post-market phase and post-market surveillance need to re-considered.
Regulators are aiming to converge their regulatory requirements for ML/MDs. Most recently, the International Medical Device Regulators Forum (IMDRF) published Machine Learning-enabled Medical Devices: Key Terms and Definitions (IMDRF 2022). A new IMDRF item is proposed for Good Machine Learning Practice (GMLP) to provide internationally harmonized principles to help promote the development of safe and effective ML/MDs.
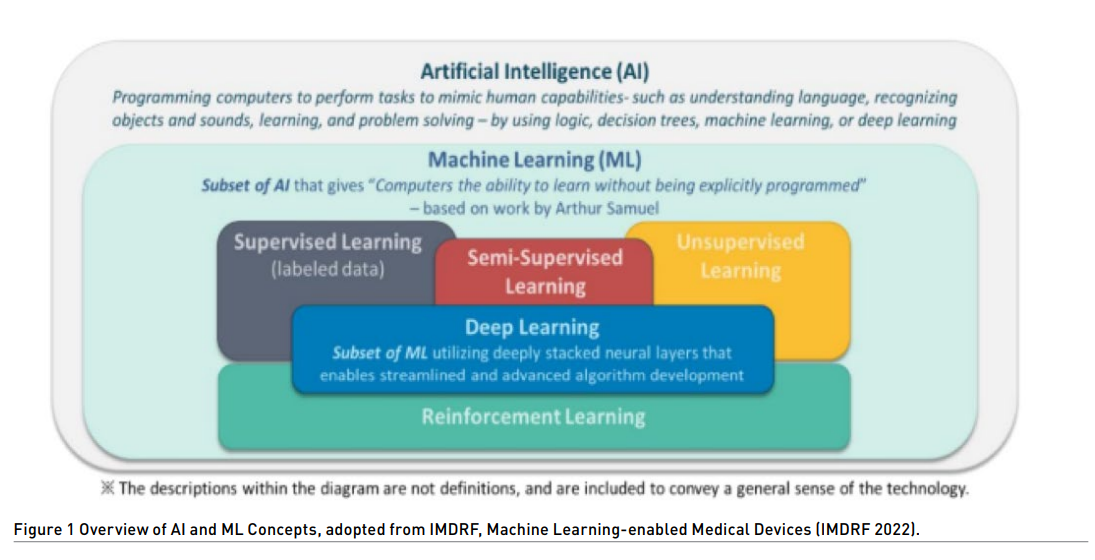
ML/MDs have the potential to address poor access to quality services for prevention, treatment and care, and lack of specially trained health personnel by leap-frogging current medical devices that require visual interpretation of results.
This has been applied for medical imaging, breast cancer screening and gastrointestinal endoscopy. Through its various departments, WHO has developed guidance for how ML/MDs might be used for cervical cancer screening and computer-aided diagnosis of tuberculosis (WH0 2021; WH0 2021) WHO guidance is forthcoming on regulatory concepts on artificial intelligence for health (Naher et al. 2023; 0ala et al. 2021).
Conclusion
ML/MDs are recognised as having the potential to improve health outcomes in settings where limited resources and a shortage of specialised healthcare professionals limit access to medical devices. Once any device with machine learning is used for a medical purpose, it becomes a medical device and therefore falls within the purview of the medical device regulatory system. Anticipating the need for refinement of existing regulations to handle the nuances for ML/MDs, WHO aims to provide guidance and support to its Member States on how to assure the quality of ML/MDs and monitor their quality, safety and performance throughout the life cycle.
Conflict of Interest
None.
References:
International Medical Device Regulators Forum [IMDRF] (2020) Adverse Event Terminology. Available from https://www.imdrf.org/working-groups/adverse-event-terminology
International Medical Device Regulators Forum [IMDRF] (2022) a Machine Learning-enabled Medical Devices: Key Terms and Definitions. Available from: https://www.imdrf.org/documents/machine-learning-enabled-medical-devices-key-terms-and-definitions
ISO. Medical devices — Post-market surveillance for manufacturers. Available from https://www.iso.org/standard/67942.html
Näher AF et al. (2023) Secondary data for global health digitalisation. Lancet Digital Health. 5 (2):e93-e101
Oala L et al. (2021) Machine Learning for Health: Algorithm Auditing & Quality Control. Journal of Medical Systems. 45, 105
The Coalition of Interested Parties (CIP) network. Available from https://cip-network-rss.org/en
World Health Organization (2019) Delivering quality-assured medical products for all 2019-2023: WHO's five-year plan to help build effective and efficient regulatory systems. World Health Organization. Available from: https://apps.who.int/iris/handle/10665/332461
World Health Organisation Executive Board (2019) Standardization of medical devices nomenclature, EB145/3 145th session. Available from: https://apps.who.int/gb/ebwha/pdf_files/EB145/B145_3-en.pdf
World Health Organization (2020) Guidance for post-market surveillance and market surveillance of medical devices, including in vitro diagnostics. World Health Organization. Available from https://apps.who.int/iris/handle/10665/337551
World Health Oragnization (2021a) Generating Evidence for Artificial Intelligence Based Medical Devices: A Framework for Training Validation and Evaluation. Geneva: World Health Oragnisation Available from https://www.who.int/publications/i/item/9789240038462
World Health Organisation (2021b) WHO consolidated guidelines on tuberculosis: module 2: screening: systematic screening for tuberculosis disease. Geneva: World Health Organisation. Available from https://www.who.int/publications/i/item/9789240022676
World Health Organization (2021c) WHO Expert Committee on Specifications for Pharmaceutical Preparations: Fifty-fifth report. Annex 10 and 11. Geneva: World Health Organization
World Health Organization (a). Prequalification of Medical Products (IVDs, Medicines, Vaccines and Immunization Devices, Vector Control): IVDs: WHO Public Reports for In Vitro Diagnostics. Available from https://extranet.who.int/pqweb/vitro-diagnostics/prequalification-reports/whopr
World Health Organization (b). Regulation and Prequalification. Available from https://www.who.int/teams/regulation-prequalification/incidents-and-SF/safety-information-for-medical-devices-including-in-vitro-diagnostics
World Health Organization (c). The WHO Global Benchmarking Tool Plus Medical Devices (GBT+MD) Revision VI+MD version. Available from https://www.who.int/tools/global-benchmarking-tools/evaluation-of-national-regulatory-systems-of-medical-devices-in-vitro-diagnostics
World Health Organization (d). WHO Global Surveillance and Monitoring System. Available from https://www.who.int/who-global-surveillance-and-monitoring-system


