




ICU Management & Practice, Volume 18 - Issue 1, 2018
 PRINT OPTIMISED
PRINT OPTIMISED
CO2 exerts potent effects on lung biology that could be clinically relevant in critically ill patients, in particular those with acute respiratory distress syndrome (ARDS). The impact of hypercapnia on outcome in these patients is yet to be determined.
The physiological effects of hypercapnia are increasingly well understood, but the literature remains confusing. Important insights have emerged regarding the impact of hypercapnia on cellular and molecular function. Hypercapnia may have potentially beneficial effects in patients with acute respiratory distress syndrome (ARDS), independent of the benefits from ventilation with low tidal volumes (Broccard et al. 2001; Curley and Laffey 2013), including reduction in pulmonary inflammation (Takeshita et al. 2003) as well as reduced oxidative alveolar damage (Shibata et al. 1998). It has also been suggested that CO2 can act as a signalling molecule via pH-independent mechanisms resulting in deleterious effects in the lung, all derived from hypercapnia (Briva et al. 2007). Clearly, hypercapnia has potent but potentially beneficial as well as potentially harmful effects. It becomes increasingly important to determine the net effect in specific conditions (Kregenow et al. 2006).
We review the biological and clinical effects of hypercapnia, especially in the patient with ARDS.
Biological effects
Ventilator-induced lung injury (VILI)
Hypercapnia has potential beneficial effects, which have been observed in experimental studies of acute lung injury, such as reduction in the levels of inflammatory mediators or oxidative alveolar damage. However, several studies suggest that CO2 could exert deleterious effects on the lung, independent of pH levels. The effects of CO2 on lung tissue are summarised in Table 1.
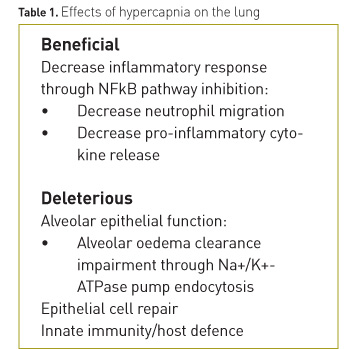
Beneficial effects
Several studies have shown that hypercapnia reduces ventilation–induced damage through its effects on mechanical stretch–induced injury:- Improvement of oxygenation, pulmonary elastance and vascular permeability with histological improvement of lung lesions (Halbertsma et al. 2010; Peltekova et al.2010).
- Prevention of the activation of the mitogen-activated protein (MAP)-kinases pathway, thus reducing the production of pro-inflammatory mediators (Gillespie and Walker 2001; Pugin 2003; Otulakowski 2014).
- Reduction of apoptosis, oxidative stress and markers of inflammation by inhibiting the activation of the MAP-kinase and stress-activated protein kinases (SAPK)/Jun amino-terminal kinases (JNK) pathways at the level of the alveolar epithelial cells (Yang et al. 2013).
- Decreased inflammatory response and improvement of pulmonary mechanics by inhibiting the canonical nuclear factor (NF)-κB pathway, degradation of IkB-α and translocation of nuclear p65 into cells (Contreras 2012).
Harmful effects
- Delayed repair of the alveolar membrane by decreasing cellular migration dependent on the NF-κB pathway (Doerr 2005; O’Toole 2009).
- Decreased clearance of alveolar oedema through inhibition of the Na+ -K+ -ATPase pump through an endocytosis process. This process has been shown to be pH independent (Briva 2007; Welch 2010; Vadász 2012; Lecuona 2013).
- Suppression of the innate immunity and host defence by inhibiting mRNA and the expressions of inflammatory cytokines (IL-6 and TNF-α) and autophagy in macrophages (Lang 2005; Oliver 2012).
Effects of hypercapnia in the setting of ARDS
Permissive hypercapnia
Hickling et al. were the first to propose protective ventilation strategies as a rescue therapy for patients with severe ARDS with the aim of limiting VILI (Hickling et al. 1990). These strategies incorporated the following measures: 1) low peak inspiratory pressure and low Vt ventilation; 2) use of positive end-expiratory pressure (PEEP); and 3) acceptance of higher partial pressure of arterial carbon dioxide (PaCO2) levels. Although that study had a series of limitations, the significant difference in hospital mortality in favour of protective ventilation strategies and permissive hypercapnia (16% vs. 39.6%) led to a series of prospective studies of protective ventilation in patients with ARDS. Based on those findings, five prospective randomised clinical trials of protective ventilatory strategies were carried out (Amato et al. 1998; Brochard et al. 1998; Stewart et al. 1998; Brower et al. 1999; Acute Respiratory Distress Syndrome Network 2000). In these studies, two showed a significant reduction in mortality (Acute Respiratory Distress Syndrome Network 2000; Brower et al. 1999) (Table 2) of protective ventilation over ventilation with high Vt (12 ml/kg/IBW). Although permissive hypercapnia was present in these studies, there are certain limitations to conclude a protective effect of CO2, such as high statistical variability, and non-randomisation of patients to receive normocapnia or hypercapnia, since the primary aim of these studies was to investigate the effect of low stretch ventilation in ALI/ARDS.
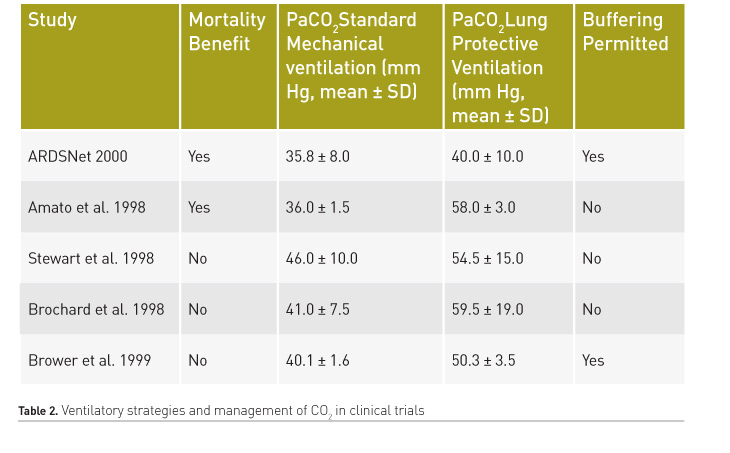
With the intention to determine whether, in addition to the effect of tidal volume reduction, there may be an independent effect of hypercapnic acidosis, the database of the ARMA trial was re–analysed (Kregenow et al. 2006). It was found that permissive hypercapnia reduced mortality in patients randomised to the higher tidal volume, but not in those receiving lower tidal volumes.
Although the approach of permissive hypercapnia is tempting, high levels of CO2 are not an easy partner for a patient with ARDS who suffers with low compliance, severe hypoxia, dyspnoea and high respiratory drive, and who requires a certain amount of sedation to allow the ventilator to take over their ventilatory distress.
Recently, in a secondary analysis of three prospective non-interventional cohort studies focusing on ARDS patients in 40 countries including a total of 1899 patients, it was found that severe hypercapnia (PaCO2 > 50 mmHg) was associated with higher ICU mortality compared to patients who kept normocapnia (OR 1.58, CI 95% 1.04–2.41; p = 0.032). Acidosis or the combination of hypercapnia and acidosis were independently positively associated with ICU mortality, as well as barotrauma, renal and cardiovascular dysfunction (Nin et al. 2017).
These findings are in line with those reported by Tiruvoipati et al. (2017). They performed a retrospective analysis including more than 250,000 patients receiving mechanical ventilation. They found that patients who developed hypercapnic acidosis (pH< 7.35 PaCO2 > 65 mmHg) during the first 24 hours of mechanical ventilation had a significantly higher mortality than those who had compensated hypercapnia or normocapnia.
Alveolar dead space
In ARDS, the alveolar dead space (VDALV) is particularly interesting. VDALV comes from respiratory units that receive disproportionately low perfusion compared with ventilation (Q < V), resulting in an increase in West Zone 1 physiology. In many patients with ARDS, the disordered pulmonary ventilation-perfusion ratio results from endothelial injury, microvascular plugging with cellular aggregates and thrombi, and disordered pulmonary blood flow, leading to increased alveolar dead space (Tomashefski et al. 1983), decreasing CO2 clearance. Clinical interest in physiological dead space measurement was reawakened in 2002, linking dead space measurements to prognosis in ARDS (Nuckton et al. 2002). Physiological dead space was measured in 179 mechanically ventilated ARDS patients on the day of the syndrome onset. The mean dead space fraction (VD/VT) was 0.54 in eventual survivors and 0.63 in patients who succumbed to the syndrome, and the risk of death increased with every 0.05 increment in VD/VT. The physiological dead space measurement outperformed all of the previous prognostic measures including traditional measures of oxygenation impairment, lung compliance and illness severity (Nuckton et al. 2002). Unfortunately, in clinical practice, VD/VT measurements are uncommon. We believe that is due to poor understanding of dead space physiology and poor integration of CO2 waveforms and derived data with other monitoring systems. Based on the arterial CO2 measurements and predicted mixed expired CO2 concentrations, Siddiki et al. found that at both day 1 and day 3 of ARDS diagnosis, patients with a VD/VT in excess of 0.50 had a risk of death that increased with every additional 0.10 increment in VD/VT (Siddiki et al. 2010), a risk prediction that almost exactly equalled the predictive power of the complete CO2 measurements on ARDS patients described by Nuckton.
Mechanical ventilation: a life-saving procedure that can kill the lung
In the past 20 years, VILI has become one of the most studied topics in critical care (Tremblay and Slutsky 2006), confirming that mechanical ventilation not only damages lung tissue but even contributes to mortality. A major cause of VILI in the past was ventilation with high Vt, which was used to maintain adequate alveolar recruitment and mean airway pressures (Pontoppidan and Geffin 1972). Although the seminal ARDS Network trial gave the guidelines for ventilation with low Vt, it has been observed that up to 30% of patients with ARDS ventilated with Vt 6 ml/kg (IBW) show alveolar overdistension, generating VILI (Terragni et al 2007). It is worth noting other mechanical ventilator-associated complications such as ventilator-associated pneumonia (VAP), ventilator-associated diaphragmatic dysfunction (VIDD), and a range of neurological disorders associated with prolonged sedation and immobilisation (Melsen and Rovers 2009; Jackson et al. 2011; Jaber et al. 2011).
Extracorporeal carbon dioxide removal: a bright future?
The use of extracorporeal devices to remove CO2 (ECCO2R) has been evaluated as an adjuvant for protective ventilation, with the aim of reducing Vt levels to values lower than 6 ml/kg (IBW). This strategy is called "ultraprotective ventilation".
Terragni et al., in a study with 32 patients with early ARDS (<72 hours), observed a decrease in the levels of inflammatory cytokines in bronchoalveolar lavage in patients undergoing ultraprotective pulmonary ventilation (Vt close to 4 ml/Kg IBW) + ECCO2R, showing less damage induced by mechanical ventilation (Terragni et al. 2009).
The Xtravent study did not observe an impact on mortality in patients with ARDS undergoing ultraprotective ventilation + ECCO2R. However, a post hoc analysis of the group of patients with PaO2/FiO2<150 showed a decrease in mechanical ventilation days in patients who received ultraprotective ventilation (Vt 3 ml/kg IBW + ECCO2R) (Bein et al. 2013).
Recently the EuroELSO working group carried out a systematic review of current clinical experience with extracorporeal CO2 removal in the critically ill (Taccone et al. 2017). They included only studies with a proper control group (studies comparing extracorporeal CO2 removal to standard treatment). Six case-control trials with a total of 279 adult patients (142 treated with ECCO2R) were identified: three of them were performed in COPD patients with hypercapnic respiratory failure and three in ARDS patients; only two trials were randomised, both in ARDS patients, in which ECCO2R was applied to allow ultraprotective ventilation. No study was sufficiently powered to disclose an effect on relevant clinical outcomes such as ICU length of stay or mortality. The overall quality of the studies was low, with a high methodological bias, not allowing any conclusion to be drawn on the clinical effectiveness of ECCO2R in critically ill patients.
The Strategy of UltraProtective Lung Ventilation With Extracorporeal CO2 Removal for New-Onset Moderate to seVere ARDS (SUPERNOVA) trial (clinicaltrials.gov/ct2/show/NCT02282657), which has completed its first pilot recruitment of patients with moderate ARDS undergoing ultraprotective ventilation + ECCO2R, will show more data on the use of extracorporeal CO2 removal in this group of patients. In addition, a randomised clinical trial, pRotective vEntilation With Veno-venouS Lung assisT in Respiratory Failure (REST) is underway to observe 90-day mortality in patients with hypoxaemic acute respiratory failure who undergo ultraprotective ventilation with ECCO2R (clinicaltrials.gov/ct2/show/NCT02654327).
To date, the available literature does not have enough evidence to make clear recommendations for the use of this technique in the critical patient, and its use is currently experimental.
Should we use a buffer to treat acidosis?
The use of a buffer to treat hypercapnic acidosis remains a common clinical practice, although it is controversial.
The justification for its use is the physiological effects associated with extreme levels of hypercapnic and metabolic acidosis (pH < 7.10) (Forsythe and Schimdt 2000; Kraut and Madias 2010). In particular these are myocardial contractility depression and haemodynamic instability refractory to catecholamine infusion, in addition to the effects of acidosis on the central nervous system and the immune function, as well as metabolism reduction.
Specific concerns exist regarding sodium bicarbonate (NaHCO3), the buffer used most frequently in the clinical setting. Although the physiochemical effect of NaHCO3 is to increase the strong ion difference, the net effect is the generation of CO2, therefore NaHCO3 is an inappropriate therapy in patients with hypercapnic acidosis.
Tromethamine (THAM: tris-hydroxy-metylaminomethane) has been considered a better choice of buffer. THAM, by diffusing easily into cells, corrects pH and simultaneously reduces carbon dioxide levels. By increasing pH levels, THAM could mitigate the adverse effects of acidosis produced on the cardiovascular system and restore haemodynamic stability (Weber et al. 2000). However, it does not solve the problem of underperfused regions of the lung, which remain under severely alkalotic conditions, or control the PaCO2 in patients with high dead space (Pesenti and Patroniti 2010).
Future directions
It is increasingly recognised that CO2 is much more than a waste product of cellular metabolism. Indeed, it is a potent biological agent that exerts multiple effects. Although the protective effects of CO2 have been observed in multiple models of acute lung injury, hypercapnia also exerts meaningful harmful effects. There is a clear need for robust evidence from well–powered RCTs to determine whether hypercapnia adds to or subtracts from the benefits of ventilation with low lung volumes and distending pressures.
ECCO2R may be a promising adjuvant therapeutic strategy for the management of patients in order to achieve protective or ultra-protective ventilation in patients with ARDS without life-threatening hypoxaemia. However, difficulties in predicting the progression of disease at an early stage may limit its use in clinical practice.
It would be interesting to analyse the impact of hypercapnia on outcome not only in the different forms of ARDS (mild, moderate and severe), but also to look at the impact of CO2 in patients at low and high risk for ARDS, and how they move from different stages according to CO2 levels and VD/VT, consider the causes of hypercapnia and target the ideal PaCO2 to best balance the favourable and unfavourable biological effects of hypercapnia.
Conflict of interest
Abbreviations
ARDS acute respiratory distress syndrome
IBW ideal body weight
JNK Jun amino-terminal kinases
MAP mitogen-activated protein
NF-κB nuclear factor-κB
PaCO2 partial pressure of arterial carbon dioxide
SAPK stress-activated protein kinases
VILI ventilator-induced lung injury
Vt tidal volume
References:
Acute Respiratory Distress Syndrome Network (2000) Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N Engl J Med, 342 (18): 1301-8.
Amato MB et al. (1998) Effect of a protective-ventilation strategy on mortality in the acute respiratory distress syndrome. N Engl J Med, 338 (6): 347-54.
Bein T et al. (2013) Lower tidal volume strategy (≈3 ml/kg) combined with extracorporeal CO2 removal versus “conventional†protective ventilation (6 ml/kg) in severe ARDS: The prospective randomized Xtravent-study. Intensive Care Med, 39 (5): 847-56.
Briva A et al. (2007) High CO2 levels impair alveolar epithelial function independently of pH, PLoS One, 2 (11): e1238.
Broccard AF et al. (2001) Protective effects of hypercapnic acidosis on ventilator-induced lung injury. Am J Respir Crit Care Med, 164 (5): 802-6.
Brochard L et al. (1998) Tidal volume reduction for prevention of ventilator-induced lung injury in acute respiratory distress syndrome. Am J Respir Crit Care Med, 158 (6): 1831-8.
Brower RG et al. (1999) Prospective, randomized, controlled clinical trial comparing traditional versus reduced tidal volume ventilation in acute respiratory distress syndrome patients. Crit Care Med, 27 (8): 1492-8.
Contreras M et al. (2012) Hypercapnic acidosis attenuates ventilation-induced lung injury by a nuclear factor-kappaB-dependent mechanism. Crit Care Med, 40 (9): 2622-30.
Curley GF et al. (2013) CrossTalk proposal: there is added benefit to providing permissive hypercapnia in the treatment of ARDS. J Physiol, 591 (11): 2763-5.
Doerr CH et al. (2005) Hypercapnic acidosis impairs plasma membrane wound resealing in ventilator-injured lungs. Am J Respir Crit Care Med, 171 (12): 1371-7.
Forsythe SM et al. (2000) Sodium bicarbonate for the treatment of lactic acidosis. Chest, 117 (1): 260-7.
Gillespie PG et al. (2001) Molecular basis of mechanosensory transduction. Nature, 413 (6852) 194-202.
Halbertsma FJ et al. (2008) Hypercapnic acidosis attenuates the pulmonary innate immune response in ventilated healthy mice. Crit Care Med, 36 (8): 2403-6.
Jaber S et al. (2011) Clinical review: ventilator-induced diaphragmatic dysfunction--human studies confirm animal model findings! Crit Care, 15 (2): 206.
Jackson DL et al. (2010) A systematic review of the impact of sedation practice in the ICU on resource use, costs and patient safety.Critical Care, 14(2): R59.
Hickling KG et al. (1990) Low mortality associated with low volume pressure limited ventilation with permissive hypercapnia in severe adult respiratory distress syndrome. Intensive Care Med, 16 (6): 372-7.
Kraut JA et al. (2010) Metabolic acidosis: pathophysiology, diagnosis and management. Nat Rev Nephrol, 6 (5): 274-85.
Kregenow DA et al. (2006) Hypercapnic acidosis and mortality in acute lung injury. Crit Care Med, 34 (1): 1-7.
Lang CJ et al. (2005) Effect of CO2 on LPS-induced cytokine responses in rat alveolar macrophages. Am J Physiol Lung Cell Mol Physiol, 289(1): L96-103.
Lecuona E et al. (2013) Protein kinase A-Ialpha regulates Na,K-ATPase endocytosis in alveolar epithelial cells exposed to high CO2 concentrations. Am J Respir Cell Mol Biol, 48 (5): 626-34.
Melsen WG et al.(2009) Ventilator-associated pneumonia and mortality: a systematic review of observational studies. Critical Care Medicine, 37 (10): 2709-18.
Nin N et al. (2017) Severe hypercapnia and outcome of mechanically ventilated patients with moderate or severe acute respiratory distress syndrome. Intensive Care Med, 43 (2): 200-8.
Nuckton TJ et al. (2002) Pulmonary dead-space fraction as a risk factor for death in the acute respiratory distress syndrome. N Engl J Med, 346 (17): 1281-6.
O'Toole D et al. (2009) Hypercapnic acidosis attenuates pulmonary epithelial wound repair by an NF-kappaB dependent mechanism. Thorax, 64 (11): 976-82.
Oliver KM et al. (2012) Hypercapnia induces cleavage and nuclear localization of RelB protein, giving insight into CO2 sensing and signaling. J Biol Chem, 287 (17): 14004-11.
Otulakowski G et al. (2014) Hypercapnia attenuates ventilator induced lung injury via a disintegrin and metalloprotease-17. J Physiol, 592 (20): 4507-21.
Peltekova V et al. (2010) Hypercapnic acidosis in ventilator-induced lung injury. Intensive Care Med, 36 (5): 869-78.
Pesenti A et al. (2010) Carbon dioxide dialysis will save the lung. Crit Care Med, 38(10 Suppl): S549-54.
Pontoppidan H et al. (1972) Acute respiratory failure in the adult. N Engl J Med, 287 (14): 690-8.
Pugin J. (2003) Molecular mechanisms of lung cell activation induced by cyclic stretch. Crit Care Med, 31 (4 Suppl): S200-6.
Shibata K et al. (1998) Hypercapnic acidosis may attenuate acute lung injury by inhibition of endogenous xanthine oxidase. Am J Respir Crit Care Med, 158 (5 Pt 1): 1578-84.
Siddiki H et al. (2010) Bedside quantification of dead-space fraction using routine clinical data in patients with acute lung injury: secondary analysis of two prospective trials. Crit Care, 14 (4): R141.
Stewart TE et al. (1998) Evaluation of a ventilation strategy to prevent barotrauma in patients at high risk for acute respiratory distress syndrome. N Engl J Med, 338 (6): 355-61.
Taccone FS et al. (2017) Extracorporeal CO2 removal in critically ill patients: a systematic review, Minerva Anestesiol, 83 (7): 762-72.
Takeshita K, et al. (2003) Hypercapnic acidosis attenuates endotoxin-induced nuclear factor-[kappa]B activation. Am J Respir Cell Mol Biol, 29 (1): 124-32.
Terragni PP et al. (2009) Tidal volume lower than 6 ml/kg enhances lung protection: role of extracorporeal carbon dioxide removal. Anesthesiology, 111 (4): 826-35.
Terragni PP et al. (2007) Tidal hyperinflation during low tidal volume ventilation in acute respiratory distress syndrome. Am J Respir Crit Care Med, 175 (2): 160-6.
Tiruvoipati R et al. (2017) Effects of hypercapnia and hypercapnic acidosis on hospital mortality in mechanically ventilated patients. Crit Care Med, 45 (7): e649-56.
Tomashefski JF Jr et al. (1983) The pulmonary vascular lesions of the adult respiratory distress syndrome. Am J Pathol. 1983, 112(1): 112-26.
Tremblay LN et al. (2006) Ventilator-induced lung injury: from the bench to the bedside. Intensive Care Med, 32(1): 24-33.
Vadász I et al. (2012) Evolutionary conserved role of c-Jun-Nterminal kinase in CO(2)-induced epithelial dysfunction. PLoS One, 7 (10): e46696.
Weber T et al. (2000) Tromethamine buffer modifies the depressant effect of permissive hypercapnia on myocardial contractility in patients with acute respiratory distress syndrome. Am J Respir Crit Care Med, 162 (4 Pt 1): 1361-5.
Welch LC et al.(2010) Extracellular signal-regulated kinase (ERK) participates in the hypercapnia-induced Na,K-ATPase downregulation. FEBS Lett, 584 (18): 3985-9.
Yang WC et al. (2013) Hypercapnic acidosis confers antioxidant and anti-apoptosis effects against ventilator-induced lung injury. Lab Invest, 93 (12): 1339-49.
Amato MB et al. (1998) Effect of a protective-ventilation strategy on mortality in the acute respiratory distress syndrome. N Engl J Med, 338 (6): 347-54.
Bein T et al. (2013) Lower tidal volume strategy (≈3 ml/kg) combined with extracorporeal CO2 removal versus “conventional†protective ventilation (6 ml/kg) in severe ARDS: The prospective randomized Xtravent-study. Intensive Care Med, 39 (5): 847-56.
Briva A et al. (2007) High CO2 levels impair alveolar epithelial function independently of pH, PLoS One, 2 (11): e1238.
Broccard AF et al. (2001) Protective effects of hypercapnic acidosis on ventilator-induced lung injury. Am J Respir Crit Care Med, 164 (5): 802-6.
Brochard L et al. (1998) Tidal volume reduction for prevention of ventilator-induced lung injury in acute respiratory distress syndrome. Am J Respir Crit Care Med, 158 (6): 1831-8.
Brower RG et al. (1999) Prospective, randomized, controlled clinical trial comparing traditional versus reduced tidal volume ventilation in acute respiratory distress syndrome patients. Crit Care Med, 27 (8): 1492-8.
Contreras M et al. (2012) Hypercapnic acidosis attenuates ventilation-induced lung injury by a nuclear factor-kappaB-dependent mechanism. Crit Care Med, 40 (9): 2622-30.
Curley GF et al. (2013) CrossTalk proposal: there is added benefit to providing permissive hypercapnia in the treatment of ARDS. J Physiol, 591 (11): 2763-5.
Doerr CH et al. (2005) Hypercapnic acidosis impairs plasma membrane wound resealing in ventilator-injured lungs. Am J Respir Crit Care Med, 171 (12): 1371-7.
Forsythe SM et al. (2000) Sodium bicarbonate for the treatment of lactic acidosis. Chest, 117 (1): 260-7.
Gillespie PG et al. (2001) Molecular basis of mechanosensory transduction. Nature, 413 (6852) 194-202.
Halbertsma FJ et al. (2008) Hypercapnic acidosis attenuates the pulmonary innate immune response in ventilated healthy mice. Crit Care Med, 36 (8): 2403-6.
Jaber S et al. (2011) Clinical review: ventilator-induced diaphragmatic dysfunction--human studies confirm animal model findings! Crit Care, 15 (2): 206.
Jackson DL et al. (2010) A systematic review of the impact of sedation practice in the ICU on resource use, costs and patient safety.Critical Care, 14(2): R59.
Hickling KG et al. (1990) Low mortality associated with low volume pressure limited ventilation with permissive hypercapnia in severe adult respiratory distress syndrome. Intensive Care Med, 16 (6): 372-7.
Kraut JA et al. (2010) Metabolic acidosis: pathophysiology, diagnosis and management. Nat Rev Nephrol, 6 (5): 274-85.
Kregenow DA et al. (2006) Hypercapnic acidosis and mortality in acute lung injury. Crit Care Med, 34 (1): 1-7.
Lang CJ et al. (2005) Effect of CO2 on LPS-induced cytokine responses in rat alveolar macrophages. Am J Physiol Lung Cell Mol Physiol, 289(1): L96-103.
Lecuona E et al. (2013) Protein kinase A-Ialpha regulates Na,K-ATPase endocytosis in alveolar epithelial cells exposed to high CO2 concentrations. Am J Respir Cell Mol Biol, 48 (5): 626-34.
Melsen WG et al.(2009) Ventilator-associated pneumonia and mortality: a systematic review of observational studies. Critical Care Medicine, 37 (10): 2709-18.
Nin N et al. (2017) Severe hypercapnia and outcome of mechanically ventilated patients with moderate or severe acute respiratory distress syndrome. Intensive Care Med, 43 (2): 200-8.
Nuckton TJ et al. (2002) Pulmonary dead-space fraction as a risk factor for death in the acute respiratory distress syndrome. N Engl J Med, 346 (17): 1281-6.
O'Toole D et al. (2009) Hypercapnic acidosis attenuates pulmonary epithelial wound repair by an NF-kappaB dependent mechanism. Thorax, 64 (11): 976-82.
Oliver KM et al. (2012) Hypercapnia induces cleavage and nuclear localization of RelB protein, giving insight into CO2 sensing and signaling. J Biol Chem, 287 (17): 14004-11.
Otulakowski G et al. (2014) Hypercapnia attenuates ventilator induced lung injury via a disintegrin and metalloprotease-17. J Physiol, 592 (20): 4507-21.
Peltekova V et al. (2010) Hypercapnic acidosis in ventilator-induced lung injury. Intensive Care Med, 36 (5): 869-78.
Pesenti A et al. (2010) Carbon dioxide dialysis will save the lung. Crit Care Med, 38(10 Suppl): S549-54.
Pontoppidan H et al. (1972) Acute respiratory failure in the adult. N Engl J Med, 287 (14): 690-8.
Pugin J. (2003) Molecular mechanisms of lung cell activation induced by cyclic stretch. Crit Care Med, 31 (4 Suppl): S200-6.
Shibata K et al. (1998) Hypercapnic acidosis may attenuate acute lung injury by inhibition of endogenous xanthine oxidase. Am J Respir Crit Care Med, 158 (5 Pt 1): 1578-84.
Siddiki H et al. (2010) Bedside quantification of dead-space fraction using routine clinical data in patients with acute lung injury: secondary analysis of two prospective trials. Crit Care, 14 (4): R141.
Stewart TE et al. (1998) Evaluation of a ventilation strategy to prevent barotrauma in patients at high risk for acute respiratory distress syndrome. N Engl J Med, 338 (6): 355-61.
Taccone FS et al. (2017) Extracorporeal CO2 removal in critically ill patients: a systematic review, Minerva Anestesiol, 83 (7): 762-72.
Takeshita K, et al. (2003) Hypercapnic acidosis attenuates endotoxin-induced nuclear factor-[kappa]B activation. Am J Respir Cell Mol Biol, 29 (1): 124-32.
Terragni PP et al. (2009) Tidal volume lower than 6 ml/kg enhances lung protection: role of extracorporeal carbon dioxide removal. Anesthesiology, 111 (4): 826-35.
Terragni PP et al. (2007) Tidal hyperinflation during low tidal volume ventilation in acute respiratory distress syndrome. Am J Respir Crit Care Med, 175 (2): 160-6.
Tiruvoipati R et al. (2017) Effects of hypercapnia and hypercapnic acidosis on hospital mortality in mechanically ventilated patients. Crit Care Med, 45 (7): e649-56.
Tomashefski JF Jr et al. (1983) The pulmonary vascular lesions of the adult respiratory distress syndrome. Am J Pathol. 1983, 112(1): 112-26.
Tremblay LN et al. (2006) Ventilator-induced lung injury: from the bench to the bedside. Intensive Care Med, 32(1): 24-33.
Vadász I et al. (2012) Evolutionary conserved role of c-Jun-Nterminal kinase in CO(2)-induced epithelial dysfunction. PLoS One, 7 (10): e46696.
Weber T et al. (2000) Tromethamine buffer modifies the depressant effect of permissive hypercapnia on myocardial contractility in patients with acute respiratory distress syndrome. Am J Respir Crit Care Med, 162 (4 Pt 1): 1361-5.
Welch LC et al.(2010) Extracellular signal-regulated kinase (ERK) participates in the hypercapnia-induced Na,K-ATPase downregulation. FEBS Lett, 584 (18): 3985-9.
Yang WC et al. (2013) Hypercapnic acidosis confers antioxidant and anti-apoptosis effects against ventilator-induced lung injury. Lab Invest, 93 (12): 1339-49.


