




ICU Management & Practice, Volume 18 - Issue 3, 2018
 PRINT OPTIMISED
PRINT OPTIMISED
Mechanisms of endotoxin-induced multi-organ damage.
Endotoxin-induced sepsis remains a leading cause of mortality in intensive care units (ICUs) worldwide. Lipopolysaccharide (LPS) identification by the immune system triggers a cascade of signalling pathways, leading to the release of several cytokines and chemokines, which orchestrate the antimicrobial and inflammatory response, though causing multiorgan damage as well. Furthermore, endotoxin is involved in the alterations of the innate and adaptative immune system, which are of utmost important in the development of immune-paralysis in sepsis and may contribute to sepsis late mortality. Even if clinical studies on techniques aiming to remove endotoxin have yielded conflicting results so far, it seems that selected subgroups of patients could benefit from their use.
Sepsis remains a leading cause of mortality in ICUs worldwide (Vincent et al. 2009) and is considered a global health priority (Reinhart et al. 2017). In 2016, new definitions and criteria of sepsis, underlining the utmost importance of the non-homeostatic host response to infection in the development of this syndrome, were published (Singer et al. 2016).
Sepsis incidence is rising due to several reasons and treatment is becoming increasingly difficult because of the spreading of multidrug-resistant bacteria. The number of Gram-negative infections in ICU is progressively increasing. In 2007, 62% of the positive isolates in ICU patients were Gram-negative organisms (Vincent et al. 2009); moreover the mortality for Gram-negative bacteraemia is higher than that for Gram-positive (Cohen et al. 2004).
Endotoxins as PAMPs
Endotoxin (LPS) is probably the most important trigger of inflammatory response in Gram-negative infection. It is a three domains essential component of the cell wall of Gram-negative bacteria and it has a highly conserved structure (Opal and Gluck 2003). However, it is the ‘regulated host response’ to LPS, rather than the intrinsic properties of LPS itself, which is responsible for the potentially lethal consequence attributed to this mediator (Monti et al. 2010).
The innate immune response is the first line of defence against infections and is based on recognition of pathogens structures, termed pathogen-associated molecular patterns (PAMPs), which are vital for survival of microorganisms and have consequently remained immutable over millennia. When PAMPs, such as LPS, peptidoglycan and lipoteichoic acid of Gram-positive bacteria, fungal glucan, (Marshfield 2011) bind to the so-called pattern recognition receptors (PRRs), the proinflammatory and antimicrobial response is triggered. It is noteworthy that also host fragments altered by cellular stress are equally recognised by the PRRs as “danger” signals, termed damage-associated molecular patterns (DAMPs) (Mogensen 2009).
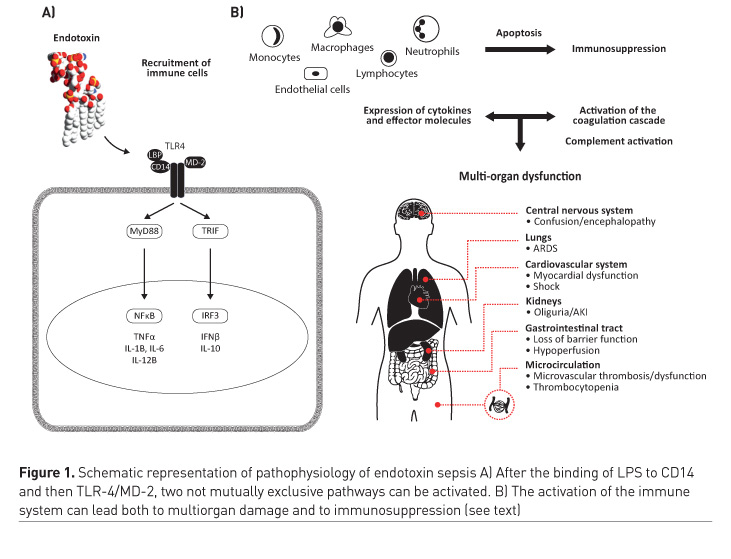
Toll-like receptors (TLRs) are the family of PRRs that have been studied more thoroughly. Currently, ten TLRs have been described. TLR-4 interacts with LPS and HSP (Opal 2010; Saha et al. 2010). LPS, through LPS binding protein, binds to the complex CD14/TLR4/MD2, which is expressed on the cell surface on both immune and non-immune cells (Molteni et al 2016). Then, two different pathways of cellular activation can occur through either the MyD88 (myeloid differentiation factor 88), which mediates the early activation of nuclear factor κB (NFκB), leading mainly to the synthesis of pro-inflammatory cytokines (TNF-α, IL1B, IL-6, IL12B), or the TRIF (Toll-like receptor domain adaptor inducing interferon-β), which, on the other hand, is involved in the late phase of transcriptional activation (IL-10) and in the development of endotoxin tolerance (Biswas and Lopez-Collazo 2009) (Figure 1A).
Clinical relevance of LPS
The most clear-cut example of the relationship between endotoxaemia and outcome is meningococcal disease (Cohen 2000). Even if this relationship is much more difficult to demonstrate in a heterogeneous ICU population, several studies have highlighted the role of endotoxaemia on progression and outcome of sepsis and septic shock (Danner et al 1991; Opal et al 1999).
In 2004, the MEDIC study, enrolling 857 ICU patients, was the first large observational cohort study to correlate endotoxin level, measured by endotoxin activity assay (EAA), with mortality. Rates of severe sepsis were 4.9%, 9.2%, and 13.2%, and ICU mortality was 10.9%, 13.2%, and 16.8% for patients with low, intermediate, and high EA levels, respectively (Marshall et al. 2004). Similarly, in a prospective study, Monti et al. (2010) showed that ‘high EA level septic shock patients’ were in need of a significantly higher vasopressors dose than intermediate and low EA groups with increased hospital mortality.
Interestingly, EA is not detectable only in patients with Gram-negative infection. More than 50% of patients admitted in ICU have intermediate or high levels of EA as compared to healthy volunteers; however, only 4% of this population had a documented Gram-negative infection (Monti et al. 2010). It has been hypothesised that the reason behind endotoxin increase in those patients is the gut barrier dysfunction (Esteban et al. 2013) associated with splanchnic hypoperfusion or gut permeability changes (McIntyre et al. 2011; Klein et al. 2007).
Pathophysiology of organ damage in Gram-negative sepsis
Endothelial dysfunction and the consequential barrier disruption leading to increased vascular permeability is critical to the pathogenesis of multi-organ failure in sepsis (Winkler et al. 2017). Specifically, stimulation of endothelial cells with LPS leads to the upregulation of several adhesion molecules (E-selectin, P-selectin, intercellular adhesion molecule-1, etc), cytokine (IFN-α, INF-γ, IL-6) and chemokine (CCL2, CCL3, CCL5). Moreover, endotoxin decreases the expression of thrombomodulin, tissue-type plasminogen activator and heparin, while increasing the expression of tissue factor (TF) and plasminogen activator inhibitor 1 (PAI-1), thus shifting the haemostatic balance from an anticoagulant to a procoagulant state. Systemic infusion of low dose LPS in healthy humans results in an enormous rise in TF mRNA levels in mononuclear cells causing thrombin generation and further haemostatic activation (Levi and Sivapalaratnam 2018). Furthermore, LPS-induced apoptosis of endothelial cells, exposing prothrombotic subendothelial proteins to clotting factors, further tilts the balance towards a procoagulant state (Seeley et al. 2012).
After an LPS challenge or sepsis insult, the heart may become dysfunctional, exhibiting a “stunning”-like profile characterised by a diffuse and reversible decrease in ejection fraction with enlargement of ventricular diameter/volumes (Chagnon et al. 2005), associated with altered muscle compliance (Chagnon et al. 2006). Microscopically, reversible and irreversible cytopathologic basic alterations include apoptosis, focal necrosis, congestion, inflammatory infiltrates, and oedema (Chagnon et al. 2006). The impairment of cardiac function during sepsis is due to several mechanisms (Flesch et al. 1999), which have not been exactly clarified yet (Yucel et al. 2017). A controversial hypothesis proposed to explain sepsis-induced cardiac dysfunctions is inadequate coronary blood flow (Chagnon et al. 2006). In fact, some studies in animals have showed that coronary blood flow is reduced by infusion of endotoxin. On the other hand, others reported a marked coronary vasodilation and even higher coronary flow in patients with sepsis (Yucel et al. 2017). Furthermore, numerous chemical mediators such as tumour necrosis factor alpha (TNF-α), MIF, interleukin-1, nitric oxide and reactive oxygen species (ROS) have been widely implicated in the pathogenesis of sepsis-induced cardiomyopathy (Chagnon et al. 2006; Yucel et al. 2017). Apoptosis seems to play an important role as well (Chagnon et al. 2005; Lancel et al. 2005). In detail, not only may endotoxin trigger heart multiple caspase activation and cytochrome c release from the mitochondria causing end-stage apoptosis of myocardial cells, but caspase-3 activation may also directly cause changes in calcium myofilament response, in troponin T cleavage, and in sarcomere disorganisation, without inducing myocardial cell death (Lancel et al. 2005). Also myocardial wall oedema per se can be an underestimated component of this reversible dysfunction altering myocardial compliance and elastance (Chagnon et al. 2006).
The frequent cardiac rhythm alteration in septic patients may be partially explained by the evidence of action potential duration (ADP)-prolongation in human pluripotent stem cell treated with LPS (Yucel et al. 2017).
LPS infusion is often used to recreate ALI (acute lung injury) in different species (Waerhaug et al. 2008). In animal models, after 1 hour from intratracheal instillation or intravenous infusion, considerable tissue injury can be observed, and it is characterised by neutrophil accumulation in the alveolar and interstitial space, alveolar wall thickening, accumulation of proteinaceous oedema and detritus in the alveolar space (Matute-Bello et al. 2011). These alterations are mostly due to the presence of profound vascular leakage causing not only movement of fluid and macro-molecules into the interstitium and airspace, but also transendothelial diapedesis of leukocytes into lung tissues, further contributing to vascular and alveolar dysfunction (Peng et al. 2004). Another important feature of ALI is the formation of microthrombi (Proudfoot et al. 2011) and alveolar fibrin deposition that, due to the extensive cross-talk between coagulation and inflammation, may further inflame the lungs (Tuinman et al. 2012). Futhermore, Rodriguez-Gonzalez et al. (2015) showed that inflammatory mediators released during LPS-induced lung epithelial cell injury might contribute to the development of septic-associated encephalopathy.
The pathogenesis of LPS-induced acute kidney injury (AKI) in humans is complex and it is not simplistically related to hypoperfusion and ischaemia (Morrell et al. 2014a; Nakano et al. 2015). In the kidney, as well as in the lung and in the heart, TLR-4 is constitutively expressed on tubular epithelial cells (especially within the apical brush border of proximal tubules) (Morrell et al. 2014a). Unfortunately, so far, little is known about the final downstream mechanisms that produce AKI after TLR-4 activation and the start of the intracellular signalling cascades (Morrell et al. 2014a). In a recent review, Morrell et al. (2014b) have suggested that the inflammatory pathway can induce renal tubular transport dysfunction with enhanced NaCl delivery to the macula densa and increased tubule-glomerular feedback, impairing the glomerular filtration rate (GFR). A study on LPS-induced AKI in mice showed LPS selectively accumulated in proximal tubule cells through a TLR-4 dependent mechanism, associated initially with a reduction in tubular flow rate and then with cells swelling and tubular obstruction (Nakano et al. 2015). Additionally, apoptosis has been proposed to play a role in the pathogenesis of septic AKI, probably through the TNFR1 (Tumor Necrosis Factor Receptor 1), as supported by a study where TNFR1−/− mice had less apoptosis in renal cells and fewer neutrophils infiltrating the kidney following LPS administration compared with TNFR1+/+ (Cunningham et al. 2002). Nonetheless, a recent study shows that LPS can also directly cause apoptosis of tubular cells through Fas-mediated and caspase-mediated pathways (Cantaluppi et al. 2008). Moreover, LPS can directly act on kidney-resident cells such as podocytes and tubular epithelium, stimulating the synthesis of inflammatory mediators (Zurovsky et al. 1995) (Figure 1B).
Endotoxin and the immune system
Due to improvements in intensive care management, early sepsis mortality has gone down during the last decades. However, late mortality is soaring. The alterations in innate and adaptive immune system induced by sepsis are thought to be of paramount importance in long-term mortality (Delano and Ward 2016).
Experimental models of sepsis have been widely used to study the so-called endotoxin tolerance, that is the desensitisation to endotoxin-induced lethality after a priming (small) dose of endotoxin before an otherwise lethal challenge dose of endotoxin. It probably occurs also in human Gram-negative sepsis (Opal 2007). The concept of ‘endotoxin tolerance’ is very helpful to identify the probable mechanisms beyond sepsis-induced alterations of the immune system and their consequences, although it is not easily adaptable to humans, because it is an oversimplification of the far more complex concept of immunoparalysis seen in human sepsis.
The mechanisms underlining LPS tolerance are still ill-understood, though recently it has been hypothesised that sepsis-induced monocyte epigenetic reprogramming may play a pivotal role in the suppressive monocyte phenotype (Delano and Ward 2016). Alterated nuclear translocation of transduction molecules, decreased stability of messenger RNA for cytokine genes (Opal 2007), and enhanced expression of two micro-RNAs (miR146 e miR155) (Biswas and Lopez-Collazo 2009) are mechanisms implicated in the genetic reprogramming of immune cells.
Altogether, after LPS challenge, monocytes produce less levels of pro-inflammatory cytokine such as TNF-α, IL-1, IL-6, IL-12 and more anti-inflammatory ones. Moreover, the ability of monocytes to present antigens is highly impaired due to reduced expression of MCH II molecules such as HLA-DR (Biswas and Lopez-Collazo 2009; Delano and Ward 2016). It is noteworthy that not only monocytes, but also dendritic cells, neutrophils, T cells (Elyce 2011), and NK cells, are involved in the genesis of immunoparalysis (Delano and Ward 2016). The widespread apoptosis of specific subsets of immune cells may contribute as well (Opal 2007) (Figure 1B).
Therapeutical approach
Since the first attempt of anti-endotoxin treatment by Ziegler, published in 1982, several strategies aiming to remove endotoxin have been proposed, from agents that inhibit endotoxin synthesis, to anti endotoxin vaccines or anti endotoxin antibodies. Unfortunately, all of them have failed the U.S. Food and Drug Administration (FDA) clinical trials (Romaschin et al. 2012). Hence, recently much research on blood purification techniques able to remove LPS has been carried out, giving contrasting results. In the fourth edition of the Surviving Sepsis Campaign Guidelines (Rhodes et al. 2017), blood purification was considered for the first time, but neither recommended in favour nor against (Ilia et al. 2017).
Several cartridges for extracorporeal blood purification have been developed, though Toraymixin® has the highest removal capacities and is the most studied. Three large randomised controlled trials (RCT)s have been set up on Polymyxin-B (PMX-B) so far, giving contrasting results. The ABDOMIX trial did not demonstrate any benefit of PMX haemoperfusion in organ failure or mortality in patients with peritonitis-induced septic shock (Payen et al. 2015), while the Early use of polymyxin B hemoperfusion in abdominal septic shock (EUPHAS) trial, reported improvement in organ dysfunction, and reduction of 28-day of mortality (Cruz et al. 2009). Similar results were demonstrated in the retrospective EUPHAS 2 registry (Cutuli et al. 2016). Finally, Evaluating the Use of Polymyxin B Hemoperfusion in a Randomized Controlled trial of Adults Treated for Endotoxemia and Septic Shock (EUPHRATES), a placebo-controlled multi-centred blinded trial was concluded in 2016 (Klein et al. 2014). Currently, the only existing report is a press release that states that PMX haemoperfusion significantly improved 28-day survival outcomes of patients with an EAA level in the range of 0.6-0.9 and a multiple organ dysfunction > 9, based on the results of a subgroup analysis (spectraldx.com/assets/spectral-rls-05.30.17.pdf).
As described by De Grooth et al. (2018), substantial between-trial heterogeneity limits the reproducibility and generalisability of septic shock research and may inhibit the discovery of beneficial therapies for specific (sub)-populations.
Conclusion
Sepsis and septic shock are still associated with a high mortality risk, and endotoxin is probably the most important trigger of inflammatory response. Although the complex interaction between the immune system and endotoxin has not been completely elucidated so far, it is clear that elevated endotoxaemia is associated with increased mortality and organ dysfunction in critically ill patients.
The clinical efficacy of extracorporeal blood purification techniques in sepsis and septic shock remains uncertain, even though Polymyxin B hemoperfusion could be considered as a complementary therapeutic strategy for unresponsive endotoxin-based septic shock.
Conflict of interest
Greta Giuliano and Gabriella Licitra declare that they have no conflicts of interest. Francesco Forfori received honoraria for lectures from Baxter, Orion, Pfizer, Biotest and Estor.
Abbreviations
ADP action potential duration
AKI acute kidney injury
ALI acute lung injury
DAMP damage-associated
molecular pattern
EAA endotoxin activity assay
GFR glomerular filtration rate
ICU intensive care unit
LPS lipopolysaccharide
PAMP pathogen-associated molecular pattern
PRR pattern recognition receptor
RCT randomised controlled trial
REA reactive oxygen species
TLR toll-like receptor
TRIF toll-like receptor domain adaptor inducing interferon-β
References:
Biswas SK, Lopez-Collazo E (2009) Endotoxin tolerance: new mechanisms, molecules and clinical significance. Trends Immunol, 30(10): 475-87.
Cantaluppi V, Assenzio B, Pasero D et al. (2008) Polymyxin-B hemoperfusion inactivates circulating proapoptotic factors. Intensive Care Med, 34(9): 1638-45.
Chagnon F, Bentourkia M, Lecomte R et al. (2006) Endotoxin-induced heart dysfunction in rats: assessment of myocardial perfusion and permeability and the role of fluid resuscitation. Crit Care Med, 34(1): 127-33.
Chagnon F, Metz CN, Bucala R et al. (2005) Endotoxin-induced myocardial dysfunction: effects of macrophage migration inhibitory factor neutralization. Circ Res, 96(10): 1095-102.
Cohen J, Cristofaro P, Carlet J et al. (2004) New method of classifying infections in critically ill patients. Crit Care Med, 32(7): 1510-26.
Cohen J (2000) The detection and interpretation of endotoxaemia. Intensive Care Med, 26 Suppl 1: S51-6.
Cruz DN, Antonelli M, Fumagalli R et al. (2009) Early use of polymyxin B hemoperfusion in abdominal septic shock: the EUPHAS randomized controlled trial. JAMA, 301(23): 2445-52.
Cunningham PN, Dyanov HM, Park P et al. (2002) Acute renal failure in endotoxemia is caused by TNF acting directly on TNF receptor-1 in kidney. J Immunol, 168(11): 5817-23.
Cutuli SL, Artigas A, Fumagalli R et al. (2016) Polymyxin-B hemoperfusion in septic patients: analysis of a multicenter registry. Ann Intensive Care, 6(1): 77.
Danner RL, Elin RJ, Hosseini JM et al. (1991) Endotoxemia in human septic shock. Chest, 99(1): 169-75.
de Grooth HJ, Postema J, Loer SA et al. (2018) Unexplained mortality differences between septic shock trials: a systematic analysis of population characteristics and control-group mortality rates. Intensive Care Med, 44(3): 311-22.
Delano MJ, Ward PA (2016) Sepsis-induced immune dysfunction: can immune therapies reduce mortality? J Clin Invest, 126(1): 23-31.
Marshfield E (2011) Endotoxin tolerance and the immune system. Plymouth Student Scientist, , 4, (2), 242-51.
Esteban E, Ferrer R, Alsina L et al. (2013) Immunomodulation in sepsis: the role of endotoxin removal by polymyxin B-immobilized cartridge. Mediators Inflamm, 2013: 50739.
Flesch M, Kilter H, Cremers B et al. (1999) Effects of endotoxin on human myocardial contractility involvement of nitric oxide and peroxynitrite. J Am Coll Cardiol, 33(4): 1062-70.
Ilia S, Briassoulis P, Briassoulis G (2017) Polymyxin B hemoperfusion in septic shock: nothing overmuch (Meden Agan)! J Thorac Dis, 9(9): 2716-9.
Klein DJ, Derzko A, Foster D et al. (2007) Daily variation in endotoxin levels is associated with increased organ failure in critically ill patients. Shock (Augusta, GA), 28(5): 524-9.
Klein DJ, Foster D, Schorr CA et al. (2014) The EUPHRATES trial (Evaluating the Use of Polymyxin B Hemoperfusion in a Randomized controlled trial of Adults Treated for Endotoxemia and Septic shock): study protocol for a randomized controlled trial. Trials, 15: 218.
Lancel S, Joulin O, Favory R et al. (2005) Ventricular myocyte caspases are directly responsible for endotoxin-induced cardiac dysfunction. Circulation, 111(20): 2596-604.
Levi M, Sivapalaratnam S (2018) Disseminated intravascular coagulation: an update on pathogenesis and diagnosis. Expert Rev Hematol, 11(8): 663-72.
Marshfield E (2011) Endotoxin intolerance and the immune system. Plymouth Student Scientist, 4(2): 242-51.
Marshall JC, Foster D, Vincent JL et al. (2004) Diagnostic and prognostic implications of endotoxemia in critical illness: Results of the MEDIC study. J Infect Dis, 190(3): 527-34.
Matute-Bello G, Downey G, Moore BB et al. (2011) An official American Thoracic Society workshop report: features and measurements of experimental acute lung injury in animals. Am J Respir Cell Mol Biol, 44(5): 725-38.
McIntyre CW, Harrison LE, Eldehni MT et al. (2011) Circulating endotoxemia: a novel factor in systemic inflammation and cardiovascular disease in chronic kidney disease. Clin J Am Soc Nephrol, 6(1): 133-41.
Mogensen TH (2009) Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev; 22(2): 240-73.
Molteni M, Gemma S, Rossetti C (2016) The Role of Toll-Like Receptor 4 in Infectious and Noninfectious Inflammation. Mediators Inflamm, 6978936.
Monti G, Bottiroli M, Pizzilli G et al. (2010) Endotoxin activity level and septic shock: a possible role for specific anti-endotoxin therapy? Contrib Nephrol, 167: 102-10.
Morrell ED, Kellum JA, Pastor-Soler NM et al. (2014a) Septic acute kidney injury: molecular mechanisms and the importance of stratification and targeting therapy. Crit Care, 18(5): 501.
Morrell ED, Kellum JA, Hallows KR et al. (2014b) Epithelial transport during septic acute kidney injury. Nephrol Dial Transplant, 29(7): 1312-9.
Nakano D, Doi K, Kitamura H et al. (2015) Reduction of tubular flow rate as a mechanism of oliguria in the early phase of endotoxemia revealed by intravital imaging. J Am Soc Nephrol, 26(12): 3035-44.
Opal SM, Gluck T (2003) Endotoxin as a drug target. Crit Care Med, 31(1 Suppl): S57-64.
Opal SM, Scannon PJ, Vincent JL et al. (1999) Relationship between plasma levels of lipopolysaccharide (LPS) and LPS-binding protein in patients with severe sepsis and septic shock. J Infect Dis, 180(5): 1584-9.
Opal SM (2010) Endotoxins and other sepsis triggers. Contrib Nephrol, 167: 14-24.
Opal SM (2007) The host response to endotoxin, antilipopolysaccharide strategies, and the management of severe sepsis. Int J Med Microbiol, 297(5): 365-77.
Payen DM, Guilhot J, Launey Y et al. (2015) Early use of polymyxin B hemoperfusion in patients with septic shock due to peritonitis: a multicenter randomized control trial. Intensive Care Med, 41(6): 975-84.
Peng X, Hassoun PM, Sammani S et al. (2004) Protective effects of sphingosine 1-phosphate in murine endotoxin-induced inflammatory lung injury. Am J Respir Crit Care Med, 169(11): 1245-51.
Proudfoot AG, McAuley DF, Griffiths MJ et al. (2011) Human models of acute lung injury. Dis Model Mech, 4(2): 145-53.
Reinhart K, Daniels R, Kissoon N et al. (2017) Recognizing sepsis as a global health priority - a WHO resolution. N Engl J Med, 377(5): 414-7.
Rhodes A, Evans LE2, Alhazzani W et al. (2017) Surviving Sepsis Campaign: International Guidelines for Management of Sepsis and Septic Shock: 2016. Intensive Care Med, 43(3): 304-77.
Rodriguez-Gonzalez R, Ramos-Nuez A, Martin-Barrasa JL et al. (2015) Endotoxin-induced lung alveolar cell injury causes brain cell damage. Exp Biol Med (Maywood), 240(1): 135-42.
Romaschin AD, Klein DJ, Marshall JC (2012) Bench-to-bedside review: clinical experience with the endotoxin activity assay. Crit Care, 16(6): 248.
Saha R, Das S, Chatterjee R et al. (2010) The pathophysiology of septic shock. Int J Pharma Bio Sciences, 1(12): 1-10.
Seeley EJ, Matthay MA, Wolters PJ (2012) Inflection points in sepsis biology: from local defense to systemic organ injury. Am J Physiol Lung Cell Mol Physiol, 303(5): L355-63.
Singer M, Deutschman CS, Seymour CW et al. (2016) The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA, 315(8): 801-10.
Tuinman PR, Dixon B, Levi M et al. (2012) Nebulized anticoagulants for acute lung injury - a systematic review of preclinical and clinical investigations. Crit Care, 16(2): R70.
Vincent JL, Rello J, Marshall J et al. (2009) International study of the prevalence and outcomes of infection in intensive care units. JAMA, 302(21): 2323-9.
Waerhaug K, Kuklin VN, Kirov MY et al. (2008) Recombinant human activated protein C attenuates endotoxin-induced lung injury in awake sheep. Crit Care, 12(4): R104.
Winkler MS, Nierhaus A, Poppe A et al. (2017) Sphingosine-1-phosphate: a potential biomarker and therapeutic target for endothelial dysfunction and sepsis? Shock, 47(6): 666-72.
Yucel G, Zhao Z, El-Battrawy I et al. (2017) Lipopolysaccharides induced inflammatory responses and electrophysiological dysfunctions in human-induced pluripotent stem cell derived cardiomyocytes. Sci Rep, 7(1): 2935.
Ziegler EJ, McCutchan JA, Fierer J et al. (1982) Treatment of gram-negative bacteremia and shock with human antiserum to a mutant Escherichia coli. N Engl J Med, 307(20): 1225-30.
Zurovsky Y, Gispaan I (1995) Antioxidants attenuate endotoxin-induced acute renal failure in rats. Am J Kid Dis, 25(1): 51-7.
Cantaluppi V, Assenzio B, Pasero D et al. (2008) Polymyxin-B hemoperfusion inactivates circulating proapoptotic factors. Intensive Care Med, 34(9): 1638-45.
Chagnon F, Bentourkia M, Lecomte R et al. (2006) Endotoxin-induced heart dysfunction in rats: assessment of myocardial perfusion and permeability and the role of fluid resuscitation. Crit Care Med, 34(1): 127-33.
Chagnon F, Metz CN, Bucala R et al. (2005) Endotoxin-induced myocardial dysfunction: effects of macrophage migration inhibitory factor neutralization. Circ Res, 96(10): 1095-102.
Cohen J, Cristofaro P, Carlet J et al. (2004) New method of classifying infections in critically ill patients. Crit Care Med, 32(7): 1510-26.
Cohen J (2000) The detection and interpretation of endotoxaemia. Intensive Care Med, 26 Suppl 1: S51-6.
Cruz DN, Antonelli M, Fumagalli R et al. (2009) Early use of polymyxin B hemoperfusion in abdominal septic shock: the EUPHAS randomized controlled trial. JAMA, 301(23): 2445-52.
Cunningham PN, Dyanov HM, Park P et al. (2002) Acute renal failure in endotoxemia is caused by TNF acting directly on TNF receptor-1 in kidney. J Immunol, 168(11): 5817-23.
Cutuli SL, Artigas A, Fumagalli R et al. (2016) Polymyxin-B hemoperfusion in septic patients: analysis of a multicenter registry. Ann Intensive Care, 6(1): 77.
Danner RL, Elin RJ, Hosseini JM et al. (1991) Endotoxemia in human septic shock. Chest, 99(1): 169-75.
de Grooth HJ, Postema J, Loer SA et al. (2018) Unexplained mortality differences between septic shock trials: a systematic analysis of population characteristics and control-group mortality rates. Intensive Care Med, 44(3): 311-22.
Delano MJ, Ward PA (2016) Sepsis-induced immune dysfunction: can immune therapies reduce mortality? J Clin Invest, 126(1): 23-31.
Marshfield E (2011) Endotoxin tolerance and the immune system. Plymouth Student Scientist, , 4, (2), 242-51.
Esteban E, Ferrer R, Alsina L et al. (2013) Immunomodulation in sepsis: the role of endotoxin removal by polymyxin B-immobilized cartridge. Mediators Inflamm, 2013: 50739.
Flesch M, Kilter H, Cremers B et al. (1999) Effects of endotoxin on human myocardial contractility involvement of nitric oxide and peroxynitrite. J Am Coll Cardiol, 33(4): 1062-70.
Ilia S, Briassoulis P, Briassoulis G (2017) Polymyxin B hemoperfusion in septic shock: nothing overmuch (Meden Agan)! J Thorac Dis, 9(9): 2716-9.
Klein DJ, Derzko A, Foster D et al. (2007) Daily variation in endotoxin levels is associated with increased organ failure in critically ill patients. Shock (Augusta, GA), 28(5): 524-9.
Klein DJ, Foster D, Schorr CA et al. (2014) The EUPHRATES trial (Evaluating the Use of Polymyxin B Hemoperfusion in a Randomized controlled trial of Adults Treated for Endotoxemia and Septic shock): study protocol for a randomized controlled trial. Trials, 15: 218.
Lancel S, Joulin O, Favory R et al. (2005) Ventricular myocyte caspases are directly responsible for endotoxin-induced cardiac dysfunction. Circulation, 111(20): 2596-604.
Levi M, Sivapalaratnam S (2018) Disseminated intravascular coagulation: an update on pathogenesis and diagnosis. Expert Rev Hematol, 11(8): 663-72.
Marshfield E (2011) Endotoxin intolerance and the immune system. Plymouth Student Scientist, 4(2): 242-51.
Marshall JC, Foster D, Vincent JL et al. (2004) Diagnostic and prognostic implications of endotoxemia in critical illness: Results of the MEDIC study. J Infect Dis, 190(3): 527-34.
Matute-Bello G, Downey G, Moore BB et al. (2011) An official American Thoracic Society workshop report: features and measurements of experimental acute lung injury in animals. Am J Respir Cell Mol Biol, 44(5): 725-38.
McIntyre CW, Harrison LE, Eldehni MT et al. (2011) Circulating endotoxemia: a novel factor in systemic inflammation and cardiovascular disease in chronic kidney disease. Clin J Am Soc Nephrol, 6(1): 133-41.
Mogensen TH (2009) Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev; 22(2): 240-73.
Molteni M, Gemma S, Rossetti C (2016) The Role of Toll-Like Receptor 4 in Infectious and Noninfectious Inflammation. Mediators Inflamm, 6978936.
Monti G, Bottiroli M, Pizzilli G et al. (2010) Endotoxin activity level and septic shock: a possible role for specific anti-endotoxin therapy? Contrib Nephrol, 167: 102-10.
Morrell ED, Kellum JA, Pastor-Soler NM et al. (2014a) Septic acute kidney injury: molecular mechanisms and the importance of stratification and targeting therapy. Crit Care, 18(5): 501.
Morrell ED, Kellum JA, Hallows KR et al. (2014b) Epithelial transport during septic acute kidney injury. Nephrol Dial Transplant, 29(7): 1312-9.
Nakano D, Doi K, Kitamura H et al. (2015) Reduction of tubular flow rate as a mechanism of oliguria in the early phase of endotoxemia revealed by intravital imaging. J Am Soc Nephrol, 26(12): 3035-44.
Opal SM, Gluck T (2003) Endotoxin as a drug target. Crit Care Med, 31(1 Suppl): S57-64.
Opal SM, Scannon PJ, Vincent JL et al. (1999) Relationship between plasma levels of lipopolysaccharide (LPS) and LPS-binding protein in patients with severe sepsis and septic shock. J Infect Dis, 180(5): 1584-9.
Opal SM (2010) Endotoxins and other sepsis triggers. Contrib Nephrol, 167: 14-24.
Opal SM (2007) The host response to endotoxin, antilipopolysaccharide strategies, and the management of severe sepsis. Int J Med Microbiol, 297(5): 365-77.
Payen DM, Guilhot J, Launey Y et al. (2015) Early use of polymyxin B hemoperfusion in patients with septic shock due to peritonitis: a multicenter randomized control trial. Intensive Care Med, 41(6): 975-84.
Peng X, Hassoun PM, Sammani S et al. (2004) Protective effects of sphingosine 1-phosphate in murine endotoxin-induced inflammatory lung injury. Am J Respir Crit Care Med, 169(11): 1245-51.
Proudfoot AG, McAuley DF, Griffiths MJ et al. (2011) Human models of acute lung injury. Dis Model Mech, 4(2): 145-53.
Reinhart K, Daniels R, Kissoon N et al. (2017) Recognizing sepsis as a global health priority - a WHO resolution. N Engl J Med, 377(5): 414-7.
Rhodes A, Evans LE2, Alhazzani W et al. (2017) Surviving Sepsis Campaign: International Guidelines for Management of Sepsis and Septic Shock: 2016. Intensive Care Med, 43(3): 304-77.
Rodriguez-Gonzalez R, Ramos-Nuez A, Martin-Barrasa JL et al. (2015) Endotoxin-induced lung alveolar cell injury causes brain cell damage. Exp Biol Med (Maywood), 240(1): 135-42.
Romaschin AD, Klein DJ, Marshall JC (2012) Bench-to-bedside review: clinical experience with the endotoxin activity assay. Crit Care, 16(6): 248.
Saha R, Das S, Chatterjee R et al. (2010) The pathophysiology of septic shock. Int J Pharma Bio Sciences, 1(12): 1-10.
Seeley EJ, Matthay MA, Wolters PJ (2012) Inflection points in sepsis biology: from local defense to systemic organ injury. Am J Physiol Lung Cell Mol Physiol, 303(5): L355-63.
Singer M, Deutschman CS, Seymour CW et al. (2016) The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA, 315(8): 801-10.
Tuinman PR, Dixon B, Levi M et al. (2012) Nebulized anticoagulants for acute lung injury - a systematic review of preclinical and clinical investigations. Crit Care, 16(2): R70.
Vincent JL, Rello J, Marshall J et al. (2009) International study of the prevalence and outcomes of infection in intensive care units. JAMA, 302(21): 2323-9.
Waerhaug K, Kuklin VN, Kirov MY et al. (2008) Recombinant human activated protein C attenuates endotoxin-induced lung injury in awake sheep. Crit Care, 12(4): R104.
Winkler MS, Nierhaus A, Poppe A et al. (2017) Sphingosine-1-phosphate: a potential biomarker and therapeutic target for endothelial dysfunction and sepsis? Shock, 47(6): 666-72.
Yucel G, Zhao Z, El-Battrawy I et al. (2017) Lipopolysaccharides induced inflammatory responses and electrophysiological dysfunctions in human-induced pluripotent stem cell derived cardiomyocytes. Sci Rep, 7(1): 2935.
Ziegler EJ, McCutchan JA, Fierer J et al. (1982) Treatment of gram-negative bacteremia and shock with human antiserum to a mutant Escherichia coli. N Engl J Med, 307(20): 1225-30.
Zurovsky Y, Gispaan I (1995) Antioxidants attenuate endotoxin-induced acute renal failure in rats. Am J Kid Dis, 25(1): 51-7.


