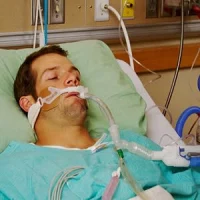
The hypothesis that mechanical ventilation (MV) can cause neurotoxicity in critically ill patients has long been debated. First reported in 1999, MV is thought to trigger chronic cognitive disorders. Brain damage induced by MV can be caused by alveolar stretching, abnormalities in PaO2/PaCO2, biotrauma and MV causing direct brain inflammation.
Alveolar stretching, induced by MV, goes on to produce systemic and cerebral mediators resulting in beta-amyloid synthesis, neuroinflammation and dopaminergic neuronal signalling. The cerebral beta-amyloid produced can cause an Alzheimer’s-like degradation of the brain. A study in Critical Care found that these outcomes can be present even after short-term MV. However, it should be noted that whilst alveolar stretching can cause adhesion molecules de novo, there is no evidence of this occurring in the brain. Also, there has been no clear translation between histopathological damage and neurocognitive impairment.
The abnormalities of PaO2/PaCO2 can lead to an increased risk of neuronal death, brain-blood flow changes and cerebral oxidative stress. It is these hypo and hyper O2 and/or CO2 molecules, which indicate MV is needed, that can remain present during MV administration. Hypoxia is the most common cause of brain damage for ARDS patients that need MV, with links to high mortality for those with acute brain damage. Also, the neuronal oxidative phosphorylation caused by this can leave patients at risk of hypoxic damage and ultimately atrophy. Hypercapnia, hyperoxia and hypocapnia can also leave patients at risk of cognitive impairment.
MV can also cause the systemic inflammation of mediators in the lungs, biotrauma, by producing molecules associated with inflammatory properties such as TNF-alpha, IL6, IL10 etc. MV can also directly influence brain inflammation through the cerebral expression of proinflammatory cytokines (e.g. TNF-alpha, IL-1beta, IL-6) and also through microglia activation. There has also been evidence of TNF-alpha levels increasing when tidal volumes have been increased. Also, increased levels of TLR-4 (Toll-like receptor 4) expression in the hippocampus after 6 hours of MV has been linked to memory impairment. The lung-brain vagal reflex, a response to MV, has also been found to activate dopamine receptors type 2 (DRD2), with evidence of it influencing apoptosis in the brain.
Methods of preventing MV-induced brain damage include the use of neuroinflammatory scavengers. For example, scavenger receptor Class A has been used for Alzheimer’s disease as it has properties which may reduce the decline of neurocognitive functions in patients. Another method is the modulation of the immune response. Studies have shown that TLR-4 antagonists have reduced neuroinflammation and the severity of cognitive disorders. Also, the selective DRD2-blocker (haloperidol) has been shown to restore the hippocampus cell survival pathways and reduce the levels of neuronal apoptosis. Other methods also include reducing the tidal volume during MV and the proper management of PaO2/PaCO2, both of which have been shown to improve neurocognitive outcomes.
It is unsure whether these outcomes are of a direct result of MV or due to the critically ill environment, however, there is still a need to retain as much neurocognitive ability in critically ill patients. In the future, the neuroinflammation of patient’s needs to be monitored during MV.
Source: Critical Care
Image Credit: iStock