




ICU Management & Practice, Volume 22 - Issue 4, 2022
 PRINT OPTIMISED
PRINT OPTIMISED
Antimicrobial Resistance in the ICU
The use of antimicrobial drugs in the hospital is very common with approximately 35% of the patients on an adult ward and up to 70% of the patients in the intensive care unit (ICU) receiving an antimicrobial drug on any given day (Versporten et al. 2018). This large antimicrobial burden exposes the patient to the risk of acquiring multidrug resistant (MDR) organisms. These MDR organisms are either the result of exogenous cross-contamination (i.e., transfer of MDR organisms from other patients or the healthcare environment) or from selection-pressure applied to the patient’s own microbiome, resulting in a competitive advantage for mutated strains (Arulkumaran et al. 2020).
ICU patients are specifically at risk for infections with MDR organisms because antimicrobial use, and therefore selection pressure, is highest in the ICU; these patients also often have advanced co-morbid illnesses and undergo invasive procedures which further exposes them to an increased risk for MDR-infections (Timsit et al. 2019). Antimicrobial resistance leads to excess deaths, prolonged hospitalisation, increased costs and the inability to perform procedures that rely on effective prophylactic antibiotic therapy (Laxminarayan et al. 2013). In response to the surge of antimicrobial resistance, antimicrobial stewardship programmes (ASP) were introduced in many hospitals around the world. The aim of an ASP programme is to improve patient outcome by ensuring optimal use of the available antimicrobial drugs. One of the core ASP interventions is antimicrobial dose-optimisation, i.e., informed decision making regarding the optimal dose and dosing regimen for the individual patient (Dyar et al. 2017; Roberts et al. 2019).
Pharmacokinetics/Pharmacodynamics of Antimicrobial Drugs
Dose-optimisation of antimicrobial drugs mainly relies on pharmacokinetic (PK) and pharmacodynamic (PD) principles. PK/PD relates the effect of the drug exposure (PK) to an outcome measurement (PD) (Nielsen and Friberg 2013). For antimicrobial drugs specifically, PK/PD describes the drug exposure necessary to achieve bacterial cell kill, while limiting toxicity and antimicrobial resistance. Three summary PK/PD indices have been defined for antimicrobial drugs. For fluoroquinolones for example, the efficacy is mainly related to area under the concentration curve of the free (ƒ) or unbound drug, inversely related to the MIC (ƒAUC/MIC). Other antimicrobial drugs, for example beta-lactam antibiotics, are considered time-dependent drugs and the PK/PD index of choice for this group is the percentage of the dosing interval the free concentration is above the MIC (ƒT>MIC). Finally, the efficacy of a third group of antibiotics, such as aminoglycosides, is best described by the peak free drug concentration inversely related to the MIC (Cmax/MIC) (Mouton et al. 2012). By convention, the magnitude of the PK/PD index necessary to achieve a certain outcome (for example a 3-log10 reduction in colony forming units (CFU/mL)) is called the PK/PD target (Nielsen and Friberg 2013).
Ideally, achieving the PK/PD target ensures a high probability of successful treatment and therefore our dosing regimens have been designed to achieve a certain predefined PK/PD target. However, a perfect dosing regimen not only ensures maximal bacterial cell kill but also tries to minimise drug toxicity and antimicrobial resistance. Unfortunately, the vast majority of preclinical PK/PD studies designed to decipher the optimal PK/PD target focused on targets linked to bacterial efficacy alone, i.e., reduction in CFU/mL, and not the drug exposure necessary to avoid antimicrobial resistance. For example, a conventional PK/PD target for intermittent infused beta-lactam antibiotics is 40-70% ƒT>MIC. Achieving this PK/PD target should ensure a 3-log10 reduction in CFU/mL after 24 hours of treatment (Dhaese et al. 2020). However, Sumi et al. (2019) have emphasised the importance of higher PK/PD targets to suppress the emergence of antimicrobial resistance (as opposed to more conventional targets to achieve a 3-log10 reduction in CFU/mL after 24 hours). For example, Tam et al. (2017) found that a Cmin/MIC (trough concentration/MIC) ratio of ≥ 3.8 (instead of 40-70% ƒT>MIC) was necessary to suppress resistance development in a 120h in vitro hollow-fibre Pseudomonas aeruginosa and Klebsiella pneumoniae infection model, although biofilm development in such an in vitro infection model may influence observations (Tam et al. 2017). Also, the majority of preclinical PK/PD experiments are only of 24-hour duration and with an initial inoculum of 105 cells (Craig 1998). Yet, clinical infections may have much higher inocula (i.e., 1010) (Feldman 1976; Wimberley et al. 1979; Low 2001) and preclinical experiments with longer treatment durations (for example 5 days instead of 24h) have clearly shown selection of mutant strains beyond the first day of treatment, even when an initial 3-log10 reduction after 24 hours was achieved (Tam et al. 2005).
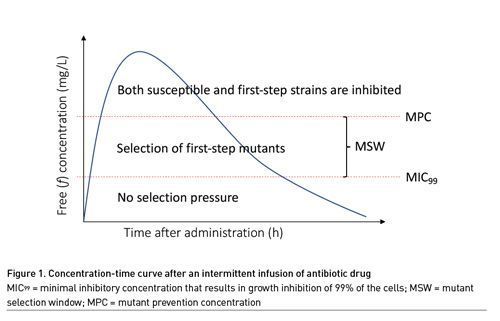
The Mutant Selection Window
Mutant prevention concentration (MPC) based indices as opposed to MIC-based indices have also been explored to describe the risk of bacterial resistance with any given dosing regimen. The MPC is the concentration that prevents growth of first-step resistant mutants. This concentration is seen as the upper limit of the mutant selection window (MSW). Above this concentration, cell growth would require two mutations. This is deemed unlikely given that the theoretical size of the inoculum with bacteria harbouring two mutations (approximately 1014) far exceeds the inocula found in clinical infections (1010) (Feldman 1976; Wimberley et al. 1979; Low 2001). The lower limit of the MSW is the lowest concentration that inhibits the growth of the majority of the drug-susceptible organisms, since below this concentration mutant strains do not have a growth advantage (Drusano et al. 2015; Firsov et al. 2003; Drlica 2003). This lower limit is approximated by the MIC99, or the minimal inhibitory concentration that results in growth inhibition of 99% of the cells. Concentrations within the MSW are expected to promote selection of resistance (Figure 1) (Drusano et al. 2015; Firsov et al. 2003).
The advantage of using the MPC instead of the MIC and denominator in the PK/PD equation is that the MPC is determined using an inoculum of 1010 instead of 105 as is common for MIC determination (Blondeau et al. 2001; Mouton et al. 2018). Using a higher inoculum has the advantage that the risk of a first-step mutant is accounted for; moreover, it also better mirrors clinical infections. However, to date, studies comparing MIC-based and MPC-based targets have not been able to clearly demonstrate superiority of one over the other (Firsov et al. 2003; Drlica 2003; Blondeau et al. 2001; Mouton et al. 2018; Olofsson et al. 2006).
Prolonged Infusion of Beta-Lactam Antibiotics and Antimicrobial Resistance
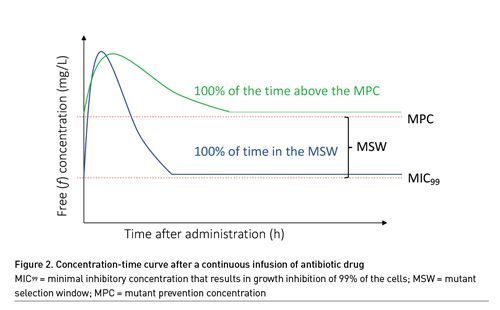
The PK of beta-lactam antibiotics is highly unpredictable in critically ill patients, mainly because of changes in kidney function and volume of distribution (Gonçalves-Pereira and Póvoa 2011). In 2013, a landmark study by Roberts et al. (2014) demonstrated that approximately 16% of the patients treated for infection with beta-lactam antibiotics administered via intermittent infusion did not achieve the PK/PD target of 50% ƒT>MIC. The observation of low target attainment rates in ICU patients fuelled the search to optimise the PK/PD of beta-lactam antibiotics in the ICU and maintaining beta-lactam antibiotic concentrations above the MIC for a prolonged period by extending the duration of infusion (i.e., prolonged infusion). The goal of prolonged infusion has always been to reduce the mortality of patients suffering from infection but little attention has been paid to differences in antimicrobial resistance with different modes of infusion (Wang et al. 2014; Lyu et al. 2017; Dulhunty et al. 2013; Abdul-Aziz et al. 2016; Bao et al. 2017; Wang 2009; Chytra et al. 2012; Vardakas et al. 2018; Roberts et al. 2016; Rhodes et al. 2018). From a theoretical point of view, continuous infusion drug concentrations may remain in the MSW for either 0 or 100% of the time, which makes it difficult to assess the impact of prolonged infusion on the risk of antimicrobial resistance (Figure 2). New acquisition, colonisation, or infection with a multi-resistant organism (MRO) is a secondary outcome of the BLING III study, a large multicentre trial with primary outcome 90-day-all-cause mortality of ICU patients receiving either intermittent or continuous infusion of piperacillin or meropenem. Study enrollment is now finished and the results are eagerly awaited (Lipman et al. 2019).
PK/PD and Antimicrobial Drug Synergism in the Treatment of Resistant Infections
PK/PD experiments have also been used to investigate synergism between two antimicrobial drugs. Synergism in PK/PD experiments is defined as a ≥ 100-fold increase in killing with the combination at 24h compared to the most active single agent and compared to the starting inoculum (Karakonstantis et al. 2022). This strategy is increasingly used to evaluate drug combinations for the treatment of infections with resistant microorganisms. For example, Lenhard et al. (2017) performed time-kill experiments with polymyxin and escalating doses of meropenem against carbapenemase-resistant Acinetobacter baumannii (CRAB). Meropenem monotherapy did not result in significant cell kill; however, in combination with polymyxin, a meropenem dose-dependent reduction in CFU/mL was seen. Synergism in this specific combination is mainly due to the mechanism of action of polymyxin which acts as a detergent making holes in the gram-negative cell wall. Synergism has also been evaluated for several other MDR organisms such as Pseudomonas aeruginosa, Staphylococcus aureus and Enterobacterales (Oh et al. 2021). Unfortunately, the evidence supporting synergism is mainly based on preclinical time-kill or PK/PD experiments and the recent European Society of Clinical Microbiology and Infectious Diseases (ESCMID) guidelines suggest the use of combination therapy for treatment of resistant organisms, although the level of evidence is low (Paul et al. 2022). Convincing in vivo data on combination of antibiotics are still missing.
Future Perspectives
In the future, dose ranging and dose fractionating studies with a clinically relevant initial inoculum (at least 107) and a clinically relevant treatment duration (at least 5 days) will be very important to determine the antibiotic exposure necessary to avoid selection of mutant strains. Based on available literature, this likely implies the need for higher PK/PD targets (or higher drug exposures). This is not without risk, given that the levels of drug toxicity for antimicrobial drugs are ill-defined. Hence, navigating on PK/PD targets for suppressing resistance alone may imply a higher risk of drug toxicity. Using a maximum tolerable dose (MTD, i.e., the highest dose possible without a risk of toxicity) may provide a more practical approach to this clinical problem (Dhaese et al. 2022). Indeed, this MTD should maximise bacterial cell kill whilst minimising the growth of first-step mutants and the adverse effects of high drug concentrations in our patients. The pitfall of this approach is the lack of strong toxicodynamic data, i.e., data describing the relationship between drug concentrations and drug toxicity. Therefore, research aimed at not only defining PK/PD targets for antimicrobial resistance but also at drug levels associated with toxicity will be paramount to optimise our current dosing regimens to suppress the emergence of resistance.
Conclusion
Pharmacokinetic/pharmacodynamic principles should aid the clinician in dose-selection, not only to improve outcomes but also prevent antimicrobial drug resistance development. However, more data are needed regarding the optimal dose exposure necessary to avoid selection of resistant strains, as well as drug levels associated with drug toxicity. Also, clinical data are urgently needed to define the role of prolonged infusions and combination therapy in infections with resistant organisms.
Conflict of Interest
None.
References:
Abdul-Aziz MH, Sulaiman H, Mat-Nor M et al. (2016) Beta-Lactam Infusion in Severe Sepsis (BLISS): A Prospective, Two-Centre, Open-Labelled Randomised Controlled Trial of Continuous versus Intermittent Beta-Lactam Infusion in Critically Ill Patients with Severe Sepsis. Intensive Care Med. 42:1535–1545.
Arulkumaran N, Routledge M, Schlebusch S et al. (2020) Antimicrobial-Associated Harm in Critical Care: A Narrative Review. Intensive Care Med. 46:225–235.
Bao H, Lv Y, Wang D et al. (2017) Clinical Outcomes of Extended versus Intermittent Administration of Piperacillin/Tazobactam for the Treatment of Hospital-Acquired Pneumonia: A Randomized Controlled Trial. Eur J Clin Microbiol Infect Dis. 36:459–466.
Blondeau JM, Zhao X, Hansen G, Drlica K (2001) Mutant Prevention Concentrations of Fluoroquinolones for Clinical Isolates of Streptococcus Pneumoniae. Antimicrob Agents Chemother. 45:433–438.
Craig WA (1998) Pharmacokinetic/Pharmacodynamic Parameters: Rationale for Antibacterial Dosing of Mice and Men. Clinical Infectious Diseases. 26:1–12.
Dhaese SAM, Hoste EA, De Waele JJ (2022) Why We May Need Higher Doses of Beta-Lactam Antibiotics: Introducing the “Maximum Tolerable Dose.” Antibiotics (Basel). 11:889.
Dhaese S, Heffernan A, Liu D et al. (2020) Prolonged Versus Intermittent Infusion of β-Lactam Antibiotics: A Systematic Review and Meta-Regression of Bacterial Killing in Preclinical Infection Models. Clin Pharmacokinet. 59:1237–1250.
Drlica K (2003) The Mutant Selection Window and Antimicrobial Resistance. Journal of Antimicrobial Chemotherapy. 52:11–17.
Drusano GL, Hope W, MacGowan A, Louie A (2015) Suppression of Emergence of Resistance in Pathogenic Bacteria: Keeping Our Powder Dry, Part 2. Antimicrob Agents Chemother. 60:1194–1201.
Dulhunty JM, Roberts JA, Davis JS et al. (2013) Continuous Infusion of Beta-Lactam Antibiotics in Severe Sepsis: A Multicenter Double-Blind, Randomized Controlled Trial. Clinical Infectious Diseases. 56, 236–244.
Dyar OJ, Huttner B, Schouten J et al. (2017) What Is Antimicrobial Stewardship? Clin Microbiol Infect. 23:793–798.
Feldman WE (1976) Concentrations of Bacteria in Cerebrospinal Fluid of Patients with Bacterial Meningitis. The Journal of Pediatrics. 88:549–552.
Firsov AA, Vostrov SN, Lubenko IY et al. (2003) In Vitro Pharmacodynamic Evaluation of the Mutant Selection Window Hypothesis Using Four Fluoroquinolones against Staphylococcus Aureus. Antimicrob Agents Chemother. 47:1604–1613.
Gonçalves-Pereira J, Póvoa P (2011) Antibiotics in Critically Ill Patients: A Systematic Review of the Pharmacokinetics of β-Lactams. Crit Care. 15:R206.
Karakonstantis S, Ioannou P, Kofteridis DD et al. (2022) In Search for a Synergistic Combination against Pandrug-Resistant A. Baumannii Methodological Considerations. Infection. 50:569–581.
Laxminarayan R, Duse A, Wattal C et al. (2013) Antibiotic Resistance—the Need for Global Solutions. The Lancet Infectious Diseases. 13:1057–1098.
Lenhard JR, Bulitta JB, Connell TD et al. (2017) High-Intensity Meropenem Combinations with Polymyxin B: New Strategies to Overcome Carbapenem Resistance in Acinetobacter Baumannii. J Antimicrob Chemother. 72:153–165.
Lipman J, Brett SJ, De Waele JJ et al. (2019) A Protocol for a Phase 3 Multicentre Randomised Controlled Trial of Continuous versus Intermittent β-Lactam Antibiotic Infusion in Critically Ill Patients with Sepsis: BLING III. Crit Care Resusc. 21:63–68.
Low DE (2001) Antimicrobial Drug Use and Resistance among Respiratory Pathogens in the Community. Clin Infect Dis. 33 Suppl 3:S206-213.
Lyu Y, Yang Y, Li X et al. (2017) Selection of Piperacillin/Tazobactam Infusion Mode Guided by SOFA Score in Cancer Patients with Hospital-Acquired Pneumonia: A Randomized Controlled Study. Ther Clin Risk Manag. 14:31–37.
Mouton JW, Muller AE, Canton R et al. (2018) MIC-Based Dose Adjustment: Facts and Fables. Journal of Antimicrobial Chemotherapy. 73:564–568.
Mouton JW, Brown DFJ, Apfalter P et al. (2012) The Role of Pharmacokinetics/Pharmacodynamics in Setting Clinical MIC Breakpoints: The EUCAST Approach. Clin Microbiol Infect. 18:E37-45.
Nielsen EI, Friberg LE (2013) Pharmacokinetic-Pharmacodynamic Modeling of Antibacterial Drugs. Pharmacol Rev. 65:1053–1090.
Olofsson SK, Marcusson LL, Komp LP et al. (2006) Selection of Ciprofloxacin Resistance in Escherichia Coli in an in Vitro Kinetic Model: Relation between Drug Exposure and Mutant Prevention Concentration. J Antimicrob Chemother. 57:1116–1121.
Oh S, Chau R, Nguyen AT, Lenhard JR (2021) Losing the Battle but Winning the War: Can Defeated Antibacterials Form Alliances to Combat Drug-Resistant Pathogens? Antibiotics. 10:646.
Paul M, Carrara E, Retamar P et al. (2022) European Society of Clinical Microbiology and Infectious Diseases (ESCMID) Guidelines for the Treatment of Infections Caused by Multidrug-Resistant Gram-Negative Bacilli (Endorsed by European Society of Intensive Care Medicine). Clinical Microbiology and Infection. 28:521–547.
Rhodes NJ, Liu J, O’Donnell JN et al. (2018) Prolonged Infusion Piperacillin-Tazobactam Decreases Mortality and Improves Outcomes in Severely Ill Patients: Results of a Systematic Review and Meta-Analysis. Critical Care Medicine. 46:236 243.
Roberts JA, Roger C, De Waele JJ (2019) Personalized Antibiotic Dosing for the Critically Ill. Intensive Care Med. 45:715–718.
Roberts JA, Abdul-Aziz M-H, Davis JS et al. (2016) Continuous versus Intermittent β-Lactam Infusion in Severe Sepsis. A Meta-Analysis of Individual Patient Data from Randomized Trials. Am J Respir Crit Care Med. 194:681–691.
Roberts JA, Paul SK, Akova M et al. (2014) DALI: Defining Antibiotic Levels in Intensive Care Unit Patients: Are Current β-Lactam Antibiotic Doses Sufficient for Critically Ill Patients? Clin Infect Dis. 58:1072–1083.
Sumi CD, Heffernan, AJ Lipman et al. (2019) What Antibiotic Exposures Are Required to Suppress the Emergence of Resistance for Gram-Negative Bacteria? A Systematic Review. Clin Pharmacokinet. 58:1407–1443.
Tam VH, Chang K.-T, Zhou J et al. (2017) Determining β-Lactam Exposure Threshold to Suppress Resistance Development in Gram-Negative Bacteria. Journal of Antimicrobial Chemotherapy. 72:1421–1428.
Tam VH, Schilling AN, Neshat S et al. (2005) Optimization of Meropenem Minimum Concentration/MIC Ratio To Suppress In Vitro Resistance of Pseudomonas Aeruginosa. Antimicrob Agents Chemother. 49:4920–4927.
Timsit J-F, Bassetti M, Cremer O et al. (2019) Rationalizing Antimicrobial Therapy in the ICU: A Narrative Review. Intensive Care Med. 45:172–189.
Vardakas KZ, Voulgaris GL, Maliaros A et al. (2018) Prolonged versus Short-Term Intravenous Infusion of Antipseudomonal β-Lactams for Patients with Sepsis: A Systematic Review and Meta-Analysis of Randomised Trials. The Lancet Infectious Diseases. 18:108 120.
Versporten A, Zarb P, Caniaux I et al. (2018) Global-PPS network Antimicrobial Consumption and Resistance in Adult Hospital Inpatients in 53 Countries: Results of an Internet-Based Global Point Prevalence Survey. Lancet Glob Health. 6, e619–e629.
Wang Z, Shan T, Liu Y et al. (2014) Comparison of 3-hour and 30-minute infusion regimens for meropenem in patients with hospital acquired pneumonia in intensive care unit: a randomized controlled clinical trial. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue. 26:644–649.
Wang D (2009) Experience with Extended-Infusion Meropenem in the Management of Ventilator-Associated Pneumonia Due to Multidrug-Resistant Acinetobacter Baumannii. International Journal of Antimicrobial Agents. 33:290–291.
Wimberley N, Faling LJ, Bartlett JG (1979) A Fiberoptic Bronchoscopy Technique to Obtain Uncontaminated Lower Airway Secretions for Bacterial Culture. Am Rev Respir Dis. 119:337–343.


