HealthManagement, Volume 16 - Issue 3, 2016
Time To Deliver
The European Commission’s ‘New Approach’ The current system for evaluating and approving new medical devices within the European Union derives from the “New Approach” first proposed by the European Commission in 1985. That legislation was designed to harmonise all industry sectors in Europe, by delegating to independent “Notified Bodies” the responsibility to confirm that new products met accepted technical standards. Although the New Approach was never planned to be appropriate for evaluating medical devices, a similar approach has been applied since the 1990s to medical technology. The Treaty of Lisbon in 2007 extended the competence of the European Union to include policies that are required to address common safety concerns in public health and achieve a high level of human health protection.
Eight years ago the European Commission responded to widespread concerns that the relevant EU Directives no longer provided an adequate basis for evaluating and approving medical devices. Serious clinical problems had occurred with some high-risk implantable medical devices that could be attributed to deficiencies in the regulatory processes. The Commission initiated a public consultation and then in September 2012 proposed two new draft EU Regulations – for the evaluation and approval of Medical Devices and In Vitro Diagnostic Devices. Around the same time, the “PIP ” scandal concerning the replacement of medical grade by industrial grade silicone in breast implants by a French manufacturer, and complications observed with metal-onmetal hip replacements, both led to an emergency “Action Plan” that has ensured that the current system is more tightly controlled.
The draft legislation made slow progress through the political channels in Brussels. Although the European Parliament approved the regulations in October 2013, after well-attended public hearings, then the experts and regulators nominated by all EU member states through the Council of the European Union considered the legislation line by line for nearly two years. Only thereafter could the “trilogue” start, through which the two EU co-legislators – the Council and the Parliament – could agree on final versions with amendments, in conjunction with the Medical Devices Unit of the European Commission acting as advisor. This process was also lengthy but it was concluded in May 2016 under the Dutch presidency of the EU. Since then, the pace of change has accelerated.
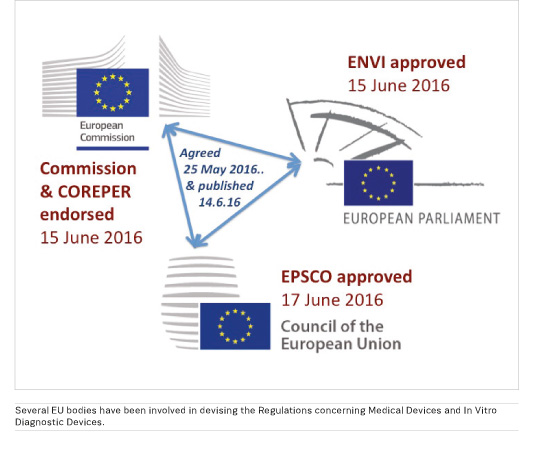
New regulations fast gaining approval
The Regulations concerning Medical Devices and In Vitro Diagnostic Devices were published on 14th June*. They have already been endorsed by the committee of permanent representatives to the European Union (COREPER); by the Environment, Public Health and Food Safety (EN VI) Committee of the European Parliament; and by the EPS CO configuration of the Council of the European Union (Employment, Social Policy, Health and Consumer Affairs). Final approvals should follow later this year, and once published in the Official Journal of the European Union the Regulations will become law. There will be a three-year period for transition, during which it will be essential for all stakeholders to implement the new systems.
The changes will affect everyone, in all branches of clinical and laboratory medicine, where modern medical practice would be impossible without medical devices. The definition of a medical device has been extended to include medical software as well as any other device (“instrument, apparatus, appliance, software, implant, reagent, material, ..”) used for the purpose of diagnosis, prevention, monitoring, prediction, prognosis, treatment or alleviation of disease. The most important changes relate to high-risk implantable medical devices (Class III ).
A major goal has been to strengthen requirements for clinical evidence concerning devices before they are approved.
The new regulation states that, for class III medical devices, manufacturers should as a general rule submit data from systematic clinical investigations that not only verify the safety and performance of the device but also establish ‘clinical benefit’ – defined as the positive impact of a device on the health of an individual, specified using meaningful, measurable, and patient-relevant clinical outcome(s). The text avoids the term “clinical effectiveness” but it will nonetheless increase the need for manufacturers to undertake more formal clinical studies. The possibility of getting new devices approved on the basis of equivalence to existing devices will decline.
At every opportunity, health care professionals have stressed that information about the basis of approval for new medical devices must be made publicly available. Now, manufacturers will need to summarise the main safety and performance aspects of all class III and implantable medical devices, and the outcome of their clinical evaluation, in documents that will be made accessible through the EU database of medical devices (Eudamed). The principle has been established that there should be adequate access to information for healthcare professionals to enable them to make informed decisions.
Commission to appoint expert panels
For the first time in Europe, the Commission will appoint expert panels in the main medical fields, which will have responsibility to assess the clinical evaluation of devices and to produce device-specific guidance, and the Commission will publish their scientific opinions and advice. A scrutiny process is also being established, by which regulators can seek assistance from these panels to evaluate individual applications from manufacturers.
Managing the new regulatory processes and ensuring that software tools are developed in time to allow efficient collaborations between national regulatory agencies will present major logistical challenges to the European Commission, and so it is planned that establishing and coordinating the expert panels will be delegated to the EU Joint Research Centre in Ispra in the north of Italy. Financial resources will be essential so that expert personnel can be recruited to deliver the new system.
For all physicians and health care professionals, it will be necessary to understand and engage with evidence-based practice concerning the use of high-risk medical devices. An essential feature of the European system, which will be strengthened, is that manufacturers must undertake comprehensive post-market surveillance; all users will need to understand that providing information from clinical follow-up is their duty, allowing some innovative products to get earlier market access while information about long-term safety and effectiveness continues to be collected. The concept of “conditional approval” has not been adopted in the legislation but it will be possible for Notified Bodies and regulatory agencies to specify conditions for vigilance relating to particular devices.
Conclusion: Deliver on changes
Health-care providers will want to link changes in regulatory practice with systems for health technology assessment, and managers will need to provide resources and systems for selecting and monitoring the devices that are used in their hospitals on the basis of the increased evidence that will be available about performance and comparative impact on clinical outcomes. For everyone, the new legislation will challenge us to reform our practices in order to ensure that patients receive the best medical devices to help them – we now have to deliver the changes that we have been campaigning for.
*http://www.emeeting.europarl.europa.eu/committees/agenda/201606/ENVI/ENVI%282016%290615_1/sitt-2571597
Key Points
- The possibility of getting new devices approved on the basis of equivalence to existing devices will decline.
- There should be adequate access to information for healthcare professionals to enable them to make informed decisions.
- New legislation will challenge us to reform our
practices in order to ensure that patients receive the best medical devices to
help them.













