



HealthManagement, Volume 19 - Issue 4, 2019
 PRINT OPTIMISED
PRINT OPTIMISED
Summary: Calcium activation and calcium sensitivity in the failing myocardium.
Historically, treatment of heart failure with inotropic agents started with the employment of extracts of foxglove, which contained a mixture of cardiac glycosides. With their low therapeutic margin due to a resulting ionic dysbalance though, cardiac glycosides have meanwhile nearly completely disappeared from the therapeutic tableau. As clinical alternatives, adrenergic agonists had been introduced, but, due to internalisation of adrenergic receptors, their therapeutic efficacy tends to vanish over time, so that at best an intermittent therapy is employed. Alternatively, phosphodiesterase inhibitors exert their effects beyond the adrenergic receptor but can impose calcium overload and energetic exhaustion of the cardiac myocyte. Thus, inotropic agents of the types of cardiac glycosides, adrenergic agonists or phosphodiesterase inhibitors have to be seen as therapeutic approaches to be taken only with great care and under strict clinical observation, not well suited for chronic medication in heart failure. As it appears, the reduced cardiac contractility in heart failure is not necessarily due to a lack of intracellular calcium ions. Therefore, in such cases, agents enhancing the sensitivity of the contractile proteins to calcium appear to be a potentially safer way to the therapy of heart failure.
You might also like: Use of levosimendan in the ICU
Introduction
Heart failure is characterised by the inability of the heart to produce sufficient blood flow through vital organs of the body. As the heart is part of the cardiovascular system, impairment of perfusion of parts of the arterial system, due to, eg atherosclerotic plaques or vasoconstriction, increase afterload and may, in the long run, affect the pumping capacity of the heart. Systemic symptoms like oedema and shortness of breath may arise and reduce the exercise capacity of the body progressively. ACE inhibitors, angiotensin receptor blockers, calcium entry blockers and the like are successfully used to reduce the afterload on the heart and can improve survival, but as exercise capacity depends critically on myocardial performance, positive inotropic agents are still needed to support the pump function of the heart.
Cardiac Glycosides for Heart Failure
The accumulation of water in oedema has historically been dubbed "Dropsy" and was successfully treated already in the 18th century by the application of extracts from the foxglove Digitalis purpurea (Withering 1785). It took, though, 168 years until the underlying mechanism of action was discovered. Schatzmann (1953) reported that cardiac glycosides inhibit the Na/K ATPase (NKA), the enzyme responsible for exchanging sodium (Na) that had entered the cardiac cell during the depolarisation phase of the action potential for potassium (K) that had left the cell during repolarisation. By this, an additional Na load is imposed on the myocardial cell. Coupled to the passive transporter (Sodium Calcium Exchanger, NCX) that exchanges intracellular Na for extracellular Calcium (Ca), an inhibition of the Na/K ATPase results therefore in a net increase in the amount of Ca ions available intracellularly for contractile activation (Figure 1, Ca fluxes 2 and 3). This results in a positive inotropic response that is principally aiding pump function, but also in an ionic imbalance that can lead to severe proarrhythmic complications.
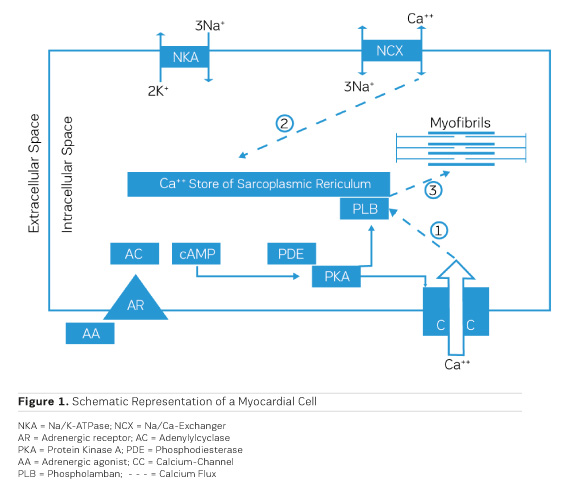
Adrenergic Agonists for Heart Failure
During bodily exercise, an increased outflow from the adrenergic system helps the heart to cope with increased circulatory needs of muscles and other organs, eg during the "fight or flight" reflex reaction. Understanding of the actions of adrenergic agonists on cardiac performance has, therefore, consequently, led to the concept of therapeutic application of adrenergic agonists in heart failure. Stimulation of adrenergic receptors triggers the production of cyclic AMP (cAMP) by adenylyl cyclase (AC) (Figure 1), which activates a cyclic AMP-dependent protein kinase (PKA) that phosphorylates a couple of proteins that are involved in handling of intracellular calcium, like the sarcolemmal calcium channel (CC) (Figure 1, Ca flux 1) and phospholamban (PLB) on the sarcoplasmic reticulum. All this together results in an increased availability of calcium ions for contractile activation and therefore a positive inotropic effect. Chronic application of adrenergic agonists, though, entails a chronically enhanced calcium activation and hence a metabolic load which the heart is not capable to cope with in the long run. The positive inotropic effect of adrenergic agonists tends, though, to fade progressively over time. The underlying cause has been identified as an intrinsic protective mechanism, namely the progressive down-regulation of adrenergic receptors by endocytosis and internalisation into the myocardial cells (for an overview, see Ferguson 2001). One way out of this dilemma is the intermittent clinical application of adrenergic agonists in chronic heart failure, which means to accept intermittent periods of no treatment (Mauro and Mauro 1986). Such observations resulted in the need for alternatives, circumventing the involvement of adrenergic receptors.
You might also like: Vasoactive medication and RCTs - An impossible marriage
Phosphodiesterase Inhibitors for Heart Failure
As shown in Figure 1, the production of cAMP in the myocardial cells is governed by adenylyl cyclase (AC), while phosphodiesterase III (PDE) cleaves cAMP. As activation of AC and production of cAMP depend critically on stimulation of the adrenergic receptor, down-regulation of the receptor leads to a loss of the inotropic effect. On the other hand, inhibition of PDE would be a therapeutic principle to protect cAMP from cleavage and is, therefore, to be expected to preserve the positive inotropic effect. At the same time, this would lead to a cAMP-dependent vasodilation in resistance vessels, thereby reducing afterload (Arnold 1993).
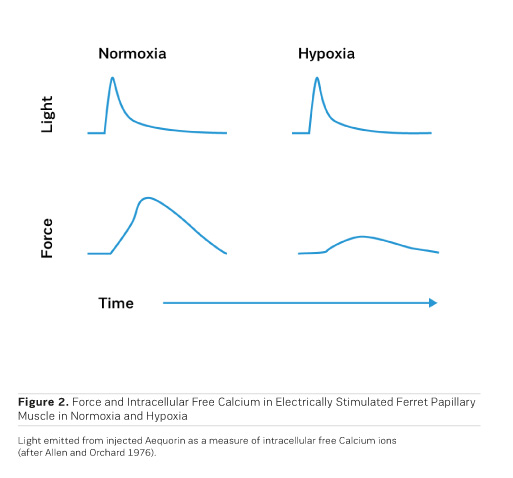
Although the beneficial effects of PDE inhibitors on cardiac performance and haemodynamics have been observed in heart failure patients, PDE inhibitors tend to negatively affect morbidity and mortality upon prolonged application (Packer et al. 1991). Like with adrenergic agonists, metabolic exhaustion and arrhythmias are certainly contributing to the observed increase in morbidity and mortality. Strikingly, cAMP, when applied to cardiac skinned fibres (Figure 3), induces a rightward shift of the calcium activation curve toward higher calcium concentrations, ie cAMP leads to a decrease in the sensitivity of the contractile structures for calcium ions through phosphorylation of Troponin I on the cardiac myofibrils (Herzig and Rüegg 1980). This at least partly explains the acceleration of diastolic relaxation in cardiac muscle under the influence of adrenergic agonists or PDE inhibitors, but it also entails a limitation of systolic force at the physiological intracellular calcium concentration of 1 μmol/l, thus this effect partly counteracts the positive inotropic action of adrenergic agonists or PDE inhibitors. Congestive heart failure is, therefore, still a disease which is clinically extremely difficult to handle. This has even led to the rather desperate judgment that trying to stimulate the failing heart with inotropic agents may be as hopeless as "flogging a dead horse." While increases in intracellular calcium certainly increase cardiac force, Allen and Orchard (1976) showed that hypoxia in intact papillary muscles leads to a loss of contractility, which is not associated with a decrease in intracellular free calcium (Figure 2). This means that in hypoxia it is not calcium that is lacking, but the sensitivity for calcium is reduced. This observation called for a search for pharmacological agents that increase the sensitivity for calcium.
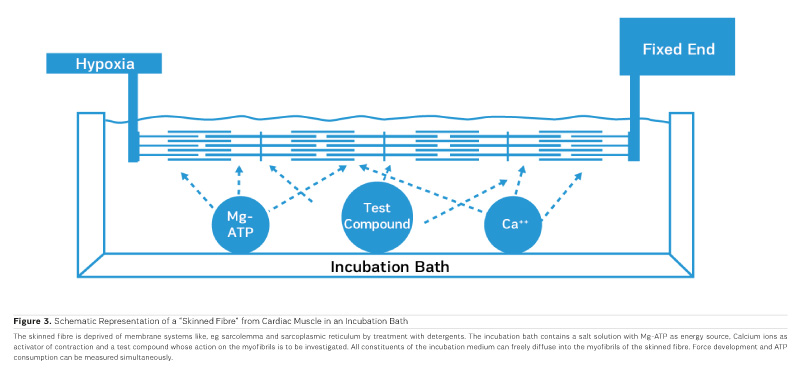
Calcium Sensitisers for Heart Failure
As PDE inhibitors have to pass through the myocardial sarcolemma to access their locus of action intracellularly (Figure 1), it appears logical to investigate whether PDE inhibitors would also exert additional actions within the myocardial cell. One elegant way to study such actions is to remove the diffusion barriers of membrane systems like the outer cell membrane (sarcolemma) and the sarcoplasmic reticulum (the intracellular storage organelle for Ca ions). This can be done in myocardial preparations by dissolving the phospholipid bilayers of these membrane systems by exposing the myocardial preparation to detergents, like Lubrol WX or Saponin. The resulting "Skinned Fibres" can then be activated by the addition of Mg-ATP as an energy source and Ca ions as "trigger" for mechanical activity (Figure 3). The skinned fibres then produce sustained contractions the amplitudes of which depend on the free Ca ion concentration in the range of 0.1 to 10 μmol/l (Figures 4 and 5).
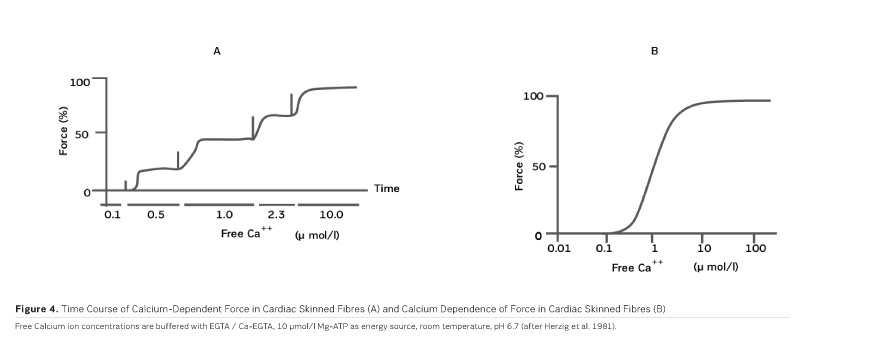
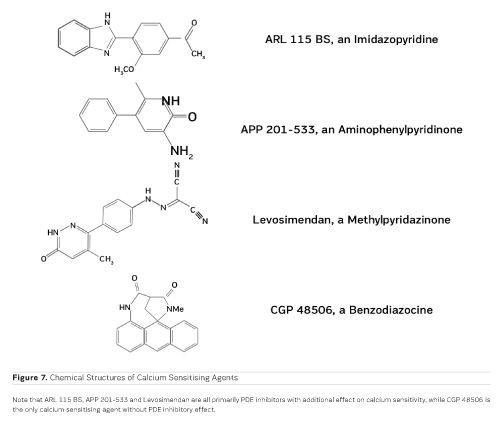
The first positive inotropic agent that was tested in such a model was ARL-115 BS, a cardiotonic PDE inhibitor (Herzig et al. 1981). As in this model membrane targets for cAMP-dependent phosphorylation are removed, and as PDE is washed out, only mechanisms that would lead to positive inotropism independent of membrane effects of the adrenergic cascade and of PDE inhibition can show up in the skinned fibre model. ARL-115 BS, in the concentration range of 350 μmol/l, leads to an increase in calcium sensitivity, ie the calcium activation curve of the skinned fibres is shifted to the left, toward lower concentrations of free calcium (Herzig et al. 1981). This observation initiated a further search for calcium sensitising agents, and the most obvious candidates for such investigations had initially been other PDE inhibitors. We (Salzmann et al. 1985) published the calcium sensitising effect of the PDE inhibitor APP 201-533 where, most importantly, we saw evidence for a concomitant effect on the economy of the contractile process, ie the calcium sensitising effect of APP 201-533 is associated with a relative reduction in the consumption of ATP by the contractile structures. This effect on cardiac economy is even more pronounced in BA 41899 and its active enantiomer CGP 48506, the first ever described calcium sensitising agent that is completely devoid of additional PDE inhibitory activity (Herold et al. 1995; Zimmermann et al. 1998) (Figures 5, 6 and 7). CGP 48506 at 10 μmol/l reduces the energy requirement from ATP for contractile activity by about 40% (Herzig unpublished). Concomitantly, systolic calcium concentrations as measured with Fluo-3 in intact papillary muscles explanted from human hearts are unaffected, while systolic shortening is increased, and diastolic relaxation is decelerated (Herzig et al. 1996). Thus, there is evidence that the calcium sensitiser CGP 48506 prolongs the attached state of the myocardial myosin cross-bridges on the actin filaments, thereby increasing the force-time-integral associated with each ATP cleavage on the contractile system, comparable to shifting the contractile system of the heart into an energy saving "overdrive."
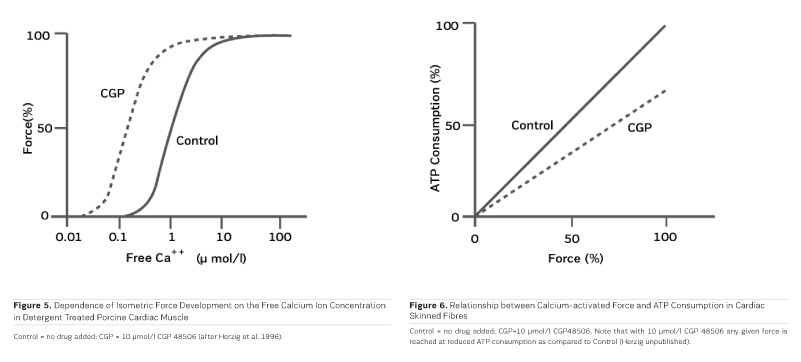
Some calcium sensitising agents have been tested clinically, but all of them are primarily PDE inhibitors with additional effects on calcium sensitivity (Kass and Solaro 2006). Levosimendan (Figure 7) has been particularly well investigated in heart failure patients. Mebazaa et al. reported in the SURVIVE Trial 2007 that levosimendan did not improve survival as compared to the adrenergic agonist dobutamine. According to Endoh (2015), the clinical observations under levosimendan can be fully explained on the basis of its PDE inhibitory effect alone. CGP 48506 (Figures 5, 6 and 7), on the other hand, is the only calcium sensitising agent without any effect on phosphodiesterase. Despite its proven efficacy as a positive inotropic agent in a variety of models, this agent has, so far, not yet been tested clinically in heart failure patients.
Conclusion
"Classical" inotropic agents of the types of cardiac glycosides, adrenergic agonists, and PDE inhibitors are acutely aiding heart failure patients, but fail in chronic application. As far as current clinical experience with those PDE inhibitors is concerned which concomitantly increase calcium sensitivity, PDE inhibition appears to, at least partially, blunt the inotropic effect of calcium sensitisation. Next to the novel myosin activators (Cleland et al. 2011), it will, therefore, be important to investigate "pure" calcium sensitisers like CGP 48506 clinically, in order to understand the true potential of the concept of calcium sensitisation, including its impact on myocardial energy demand (Salzmann et al. 1985; see also Figure 6), independent of other inotropic mechanisms. Such studies are still missing and should be undertaken with priority. With such information, inotropic agents for heart failure may, in the end, become more than just wishful thinking.
Key Points
- Mixed clinical experience with cardiac glycosides, adrenergic agonists and PDE inhibitors
- Calcium activates the heart, but is calcium lacking in heart failure?
- Don`t increase calcium, try calcium sensitising agents
References:
Allen DG, Orchard CH (1983) Intracellular calcium concentration during hypoxia and metabolic inhibition in mammalian ventricular muscle. J Physiol 339:107-122
Arnold JM (1993) The role of phosphodiesterase inhibitors in heart failure. Pharmacol Ther 57:161-170
Cleland JG, Teerlink JR, Senior R, Nivontov EM, McMurray JJ, Lang CC et al. (2011) The effects of the cardiac myosin activator, omecamtiv mecarbil, on cardiac function in systolic heart failure: a double-blind, placebo-controlled, crossover, dose-ranging phase 2 trial. Lancet 378:676-683
Endoh M (2015) Does levosimendan act as a calcium sensitizer or PDE 3 inhibitor? Br J Pharmacol 172:4594-4596
Ferguson SS (2001) Evolving concepts in G protein-coupled receptor endocytosis: The role in receptor desensitization and signaling. Pharmacol Rev 53:1-24
Herold P, Herzig JW, Wenk P, Leutert T, Zbinden P, Fuhrer W, Stutz S, Schenker K, Meier M, Rihs G (1995) 5-Methyl-6-phenyl-1,3,5,6-tetrahydro-3,6-methano-1,5-bezodiazocine-2,4-dione (BA 41899): representative of a novel class of purely calcium-sensitizing agents. J Med Chem 38:2946-2954
Herzig JW, Chiesi M, Depersin H, Grüninger S, Hasenfuss G, Kubalek R, Leutert T, Pieske B, Pioch K, Wenk P, Holubarsch C (1996) Calcium sensitization in idiopathic dilated human myocardium. Arzneim Forsch Drug Res 46:586-493
Herzig JW, Feile K, Rüegg JC (1981) Activating effects of AR-L 115 BS on the calcium sensitive force, stiffness and unloaded shortening velocity in isolated contractile structures from mammalian heart muscle. Arzneim Forsch Drug Res 31:188-191
Herzig JW, Rüegg JC (1980) Investigations on glycerinated cardiac muscle fibres in relation to the problem of regulation of cardiac contractility - effects of Calcium and cAMP. Basic Res Cardiol 75:26-33
Kass DA, Solaro RJ (2006) Mechanisms and use of calcium-sensitizing agents in heart failure. Circulation 113:305-315
Mauro VF, Mauro LS (1986) Use of intermittent dobutamine infusion in congestive heart failure. Drug Intell Clin Pharm 20, 919-924
Mebazaa A, Nieminen MS, Packer M, Cohen-Solal A, Kleber FX, Pocock SJ, Thakkar R, Padley RJ, Pöder P, Kivikko M (2007) Levosimendan vs. dobutamine for patients with acute decompensated heart failure: the SURVIVE randomized trial. JAMA 297:1883-1891
Packer M, Carver JR, Rodeheffer RJ, Ivanhoe RJ, DiBianco R, Zeldis SM, Hendrix GH, Brommer WJ, Elkayam U, Kukin ML, Sollano JA et al. (1991) Effect of oral milrinone on mortality in severe chronic heart failure. N Engl J Med 325:1468-1475
Salzmann R, Bormann G, Herzig JW, Markstein R, Scholtysik G (1985) Pharmacological actions of APP 201-533, a novel cardiotonic agent. J Cardiovasc Pharmacol 7:588-596
Schatzmann H J (1953) Cardiac glycosides as inhibitors for the active potassium and sodium transport across the red cell membrane. Helv Physiol Pharmacol Acta 11:346-354
Withering W (1785) An account of the foxglove, and some of its medical uses: with practical remarks on dropsy, and other diseases. Birmingham 1785
Zimmermann N, Boknik P, Gams E, Herzig JW, Neumann N, Scholz H (1998) Calcium sensitization as new principle of inotropic therapy in end-stage heart failure? Eur J Cardiothor Surg 14:70-75
