HealthManagement, Volume 15 - Issue 3, 2015
Autosomal dominant polycystic kidney disease (ADPKD) is an inherited, chronic, progressive condition in which cysts develop in the kidneys and other organs (Torres et al. 2007; Chapman et al. 2015; Ong et al. 2015). ADPKD is an important cause of chronic kidney disease, accounting for around one in ten of all patients needing renal replacement therapy (RRT) via dialysis or kidney transplantation (Spithoven et al. 2014a). Approximately 50,000 people with ADPKD receive RRT across Europe, at an estimated cost of €1.5 billion/year (Spithoven et al. 2014a).
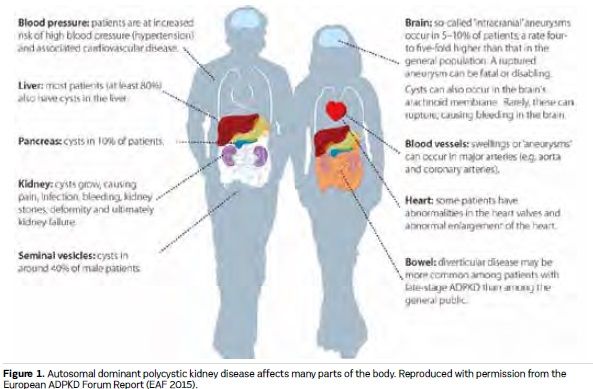
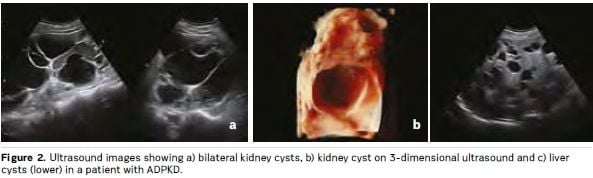
Kidney cysts develop throughout life in patients with ADPKD, causing symptoms and complications that include pain, abdominal distention, cyst infection and bleeding. Presentation and progression of ADPKD is highly variable, but on average patients commence RRT between 55 and 60 years of age (Spithoven et al. 2014b). Most patients with ADPKD also have liver cysts, and many other organs can be affected (see Figures 1 and 2). The disease has lifelong psychological effects that, together with physical manifestations, can reduce quality of life and affect work, social and family lives (Carr et al. 2014; Tong et al. 2015a; EAF 2015). Evidence suggests that many doctors may underestimate the impact that ADPKD can have on patients, even in the early stages (Carr et al. 2014). Clinical practice for ADPKD diagnosis, assessment, treatment and support varies within and between European countries. There is a lack of evidencebased consensus guidelines, standardised care pathways and treatment options, and little coordination of care policies and services. The practical enhancement of patients’ roles in decision-making, in their own care and more widely in the design and implementation of healthcare policies, systems and services, is central to efforts to address these issues (EAF 2015; Youssouf et al. 2015).
This article reviews recent developments in the field of ADPKD and highlights opportunities for patient empowerment within the context of policy-focused strategies to improve ADPKD care, recommended by an expert group called the European ADPKD Forum (EAF) (EAF 2015).
Coordinated, Multidisciplinary, Patient-Centred Care is Essential
The EAF recommends the development of tiered care approaches to ensure that patients diagnosed with ADPKD have appropriate access to specialist, multidisciplinary management according to evolving best practice, and that patient organisations should be consulted to inform decisions regarding associated health policies (EAF 2015).
An example of such an approach is a model in which all patients diagnosed with ADPKD have access to a specialist nephrology centre where multidisciplinary, patient-centred care can be provided (see Figure 3 – available in online version). Referral to specialist centres would be encouraged for early prognostic assessment, genetic testing and the investigation and management of disease manifestations and complications. The expertise of specialists in hepatology, urology, cardiology and radiology should be available according to need, together with associated genetic counselling services. The coordination of specialist care would be expected to improve the efficiency of healthcare resource utilisation, for example through a more targeted use of novel diagnostic and therapeutic interventions (EAF 2015).
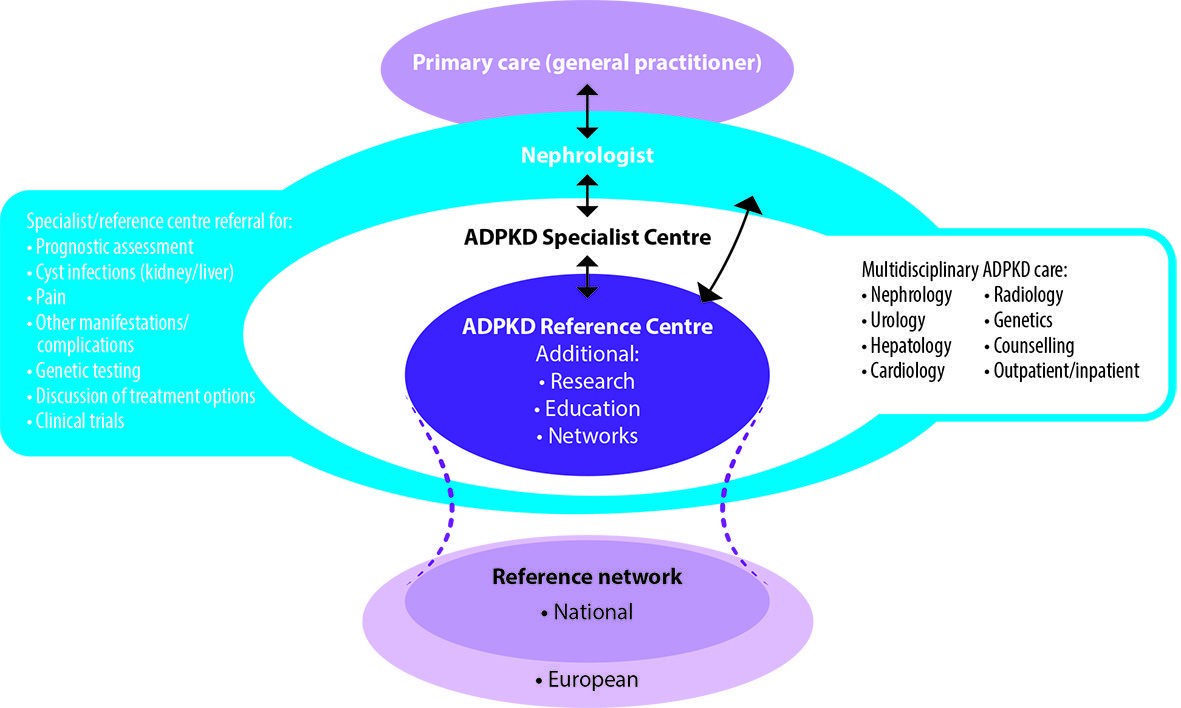
It is anticipated that some ADPKD specialist centres would be designated as Reference Centres that would undertake basic, translational and clinical research, medical education and the development and implementation of future clinical guidelines and best practice standards. The EuroCYST initiative has established a network of 14 ADPKD centres in Belgium, Czech Republic, France, Germany, Italy, Netherlands, Spain, Switzerland, Turkey and the United Kingdom (http://eurocyst. org) (Petzold et al. 2014). Funded by the European Renal Association-European Dialysis and Transplantation Association (ERA-EDTA), the project aims to recruit 1100 adult patients to be followed for at least three years in an observational cohort study. An expanded European network of ADPKD Reference Centres would further facilitate research and the establishment of harmonised, integrated, patient-centred care pathways (EAF 2015).
Patients Need to be Informed to be Empowered
Patients with progressive, lifelong diseases should be enabled to be fully active partners in their own care. Patients diagnosed with ADPKD, and their families, should be provided with reliable, simple, userfriendly, country-specific, written information on ADPKD in order to allow them to fully participate in decision-making (see Panel 1).
The UK PatientView (www.patientview. org) system, which provides patients with chronic kidney disease web-based access to their laboratory results (Woywodt et al. 2014), is an innovative approach to empowering patients that may be applicable to ADPKD. Patients’ interest in such information is supported by an analysis of usage data from over 11,000 registered patients. More than half logged on twice a month on average during follow-up periods of up to four years (Phelps et al. 2014).

A survey of almost 4,000 patients from 36 European countries by the European Kidney Patients’ Federation (EKPF) (formerly CEAPIR) found that almost twothirds (64%) did not receive or could not remember receiving education on how to manage kidney disease in their daily life (Van Biesen et al. 2014). Patients with ADPKD should also have access to advice and support to help them deal with the impact of their condition on their mental health, relationships, employment, financial affairs and health insurance. Each person with ADPKD has a 50% (ie one in two) chance that any child he or she has will inherit the gene causing the disease. Reproductive counselling is vital to explain this risk and associated issues. Patients should also be routinely referred to patient organisations, which can provide further information and support.
Managing Pain
ADPKD patients can experience acute and chronic pain, and often report that it affects their mood, sleep, relationships, daily activities and enjoyment of life (Oberdahn et al. 2014; Tong et al. 2015a). Qualitative research among patients suggests that physicians may not adequately appreciate the level of pain associated with ADPKD. Patients have reported that analgesic therapy is often inadequate, and that they do not feel sufficiently involved in decision-making regarding pain therapy (Tong et al. 2015b).
Experts have proposed a management algorithm for chronic pain in patients with ADPKD, starting with conservative nonpharmacological measures, progressing to pharmacological, minimally invasive treatments and ultimately to complex, invasive therapies where necessary (Casteleijn et al. 2014). An interdisciplinary approach should be used to manage intractable pain, involving both pain specialists and nephrologists. Patients with chronic pain should also be assessed for depression and provided with appropriate treatment and support. It is hoped that this model will serve as an example for patient-centred pain care pathways in other countries.
Reducing Cardiovascular Risk
Patients with ADPKD are at risk of high blood pressure and cardiovascular disease. Cardiovascular risk factor management has been credited with observed improvements in life expectancy in this population (Spithoven et al. 2014a). However, evidence from Spain suggests that cardiovascular risk management is often suboptimal in patients with ADPKD (Gorriz et al. 2014), and initiatives for healthcare professionals and patients are needed to promote best practice and adherence (Chapman et al. 2015).
Results from the landmark, doubleblind, randomised HALT Progression of Polycystic Kidney Disease (HALT-PKD) study have informed antihypertensive strategies in ADPKD (Schrier et al. 2014). 558 hypertensive patients with early stage ADPKD were randomised to rigorous blood pressure control (target 95–110/60–75 mmHg) or standard control (target 120–130/70–80 mmHg), and to an angiotensin-converting enzyme (ACE) inhibitor (lisinopril) plus an angiotensinreceptor blocker (ARB; telmisartan) or lisinopril plus placebo. Patients who received rigorous blood pressure control showed a 14% slower annual increase in heightadjusted total kidney volume (TKV) — a measure of ADPKD progression — than those with standard control (5.6% vs 6.6%; p=0.006). Blood pressure in patients with ADPKD can be effectively controlled by blockade of the renin-angiotensin-aldosterone system, with benefits also on leftventricular mass index and urinary albumin excretion. Dual ACE inhibitor-ARB therapy did not significantly benefit TKV versus lisinopril alone (Shrier et al. 2014).
Dialysis and Transplantation
The costs of ADPKD care increase substantially when RRT is needed. RRT generally accounts for a disproportionate amount of healthcare resource utilisation and costs for all forms of chronic kidney disease. In the UK it has been estimated that only 2% of patients with chronic kidney disease need RRT, yet RRT accounts for 54% of all spending on chronic kidney disease (Kerr et al. 2012). Optimising RRT use is important to improve the costeffectiveness of care as well as patient outcomes, especially considering that the number of patients receiving RRT for ADPKD in Europe increased by 60% between the periods of 1991–1995 and 2006–2010 (Spithoven et al. 2014a).
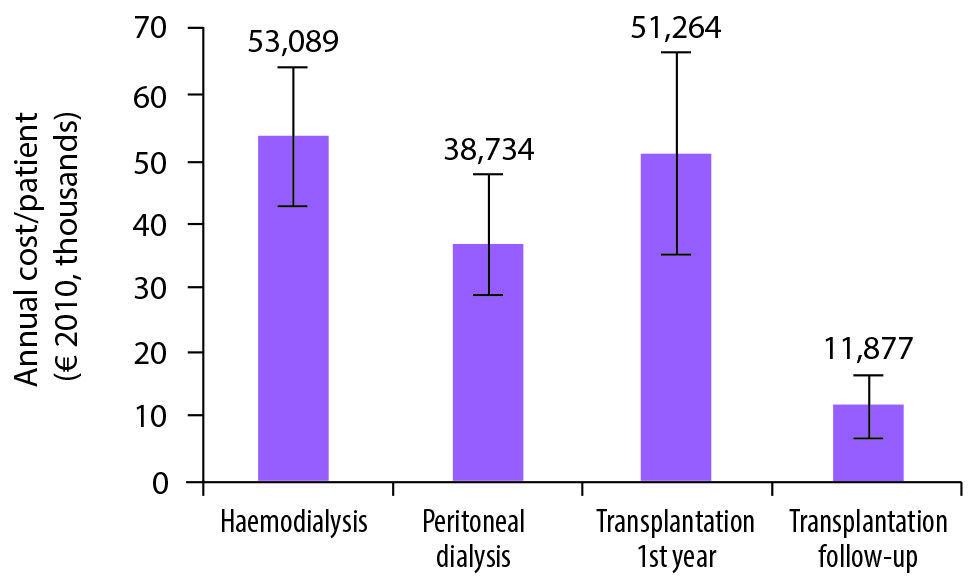
From the perspective of the healthcare system, kidney transplantation is far more cost-effective than dialysis (see Figure 4 in the online version). Haemodialysis — the most common RRT modality in patients with ADPKD — is estimated to cost €55,500 per patient per year on average across Europe. While transplantation costs are similar in the first year, the annual cost associated with transplantation follow-up thereafter is estimated at around €12,000 (Spithoven et al. 2014a). There is a clear need for further promotion of kidney transplantation, including in patients with ADPKD, as part of EU efforts in this field (EAF 2015).
Results from the survey of kidney disease patients coordinated by EKPF suggests that three-quarters had been involved in the process of choosing their RRT modality, and that patients who were involved in selection were more satisfied with their treatment. Respondents were more often satisfied with information provided on in-centre haemodialysis (90%) and transplantation (87%) than on peritoneal dialysis (79%) or home haemodialysis (61%) (Van Biesen et al. 2014). A large international survey of patients undergoing haemodialysis also revealed information provision to be among the aspects of care with which patients were least satisfied (Palmer et al. 2014).
New Approaches to Disease Modification
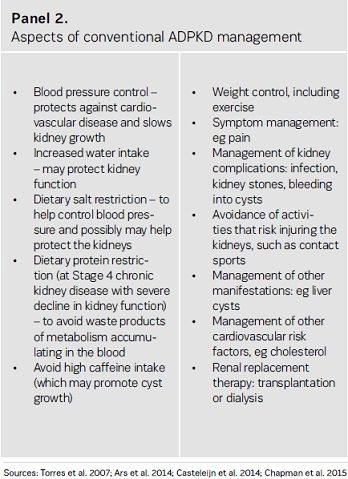
Treatments that can slow cyst development in patients with ADPKD, thereby delaying kidney failure, have the potential to reduce the need for costly and invasive dialysis or transplantation. Although a variety of approaches are conventionally taken to protect the kidneys (see Panel 2), these do not appear to be effective in delaying the need for RRT in patients with ADPKD (Spithoven et al. 2014b).
Treatments to slow ADPKD progression are urgently needed to maintain patients’ quality of life and delay the need for dialysis and transplantation. In May 2015, tolvaptan, a vasopressin-2-receptor antagonist (Torres et al. 2012), was granted market authorisation for use to slow the progression of cyst development and renal insufficiency of ADPKD in adults with stage 1 to 3 chronic kidney disease at initiation of treatment and with evidence of rapidly progressing disease. Tolvaptan treatment must be initiated and monitored under the supervision of physicians with expertise in managing ADPKD and a full understanding of the risks of tolvaptan therapy, including hepatic toxicity and monitoring requirements (Otsuka 2015). Other investigational disease-modifying agents are in development (Chang & Ong 2013; www.clinicaltrials.gov).
Patients have a vital contribution to make to the processes of medicines regulation and healthcare technology assessment (HTA). In particular, patients can provide unique insights into the disease impact, existing unmet needs and risk-benefit assessment of new medicines. The European Medicines Agency (EMA) includes patients on its Scientific Committees. However, patient involvement in HTA is highly variable. While there are examples of good practice (HTA International 2015), patients in some countries are afforded little or no input into decisions that could profoundly affect their lives. The EAF has called for HTA bodies to seek to engage patients and patient organisations in assessments according to published standards (EAF 2015).
The development and introduction of new therapies for ADPKD will necessitate the identification of patients who will benefit from treatment, in order to guide individualised treatment and monitoring, and to aid study recruitment. It has been recommended that governments support measures for routine prognostic assessment in patients with ADPKD (EAF 2015).
The progression of early ADPKD cannot be monitored by conventional measures of chronic kidney disease (eg the glomerular filtration rate). Research is ongoing to refine and validate predictive tools, based on the TKV, genetic testing and other variables, for use in clinical practice and clinical trials. ADPKD results from genetic mutations in one of two genes, PKD1 and PKD2. Mutations in PKD1 account for 85% of cases of ADPKD where a mutation is identified, and are associated with a more severe and progressive disease course. Genetic testing to identify the causative mutation can be used to confirm the diagnosis in some situations, particularly in children (Chapman et al. 2015), and for kidney donor assessment in a relative of someone with ADPKD (Sims et al. 2014). Use of genetic testing is limited in part by its complexity and cost and the need for genetic expertise. The recent development of faster and cheaper ‘nextgeneration sequencing’ tests may lead to a greater role for genetic testing both in diagnosis of ADPKD and in assessing prognosis. Standardised reporting, as well as physician education and genetic counselling for patients, will be needed to optimise the use of new tests (Chapman et al. 2015).
Defining Priorities for Research
Patients should not merely be the subjects of medical research, but be actively involved in the design, commissioning and conduct of research, and in the review and dissemination of its findings. According to a recent systematic review, only 25% of studies that elicited stakeholders’ priorities for research in kidney disease explicitly involved patients (Tong et al. 2015).
The lack of established international clinical guidelines for ADPKD management is due in part to limitations in the evidence base. A Kidney Disease Improving Global Outcomes (KDIGO) Controversies Conference Report, co-developed by healthcare professionals and patient representatives, has evaluated this evidence base and defined the outstanding research needs with a view to the development of future clinical guidelines (Chapman et al. 2015). From a patient’s perspective, there is a pressing need for the development of specific patient-reported tools to measure the psychological impact of ADPKD and studies to evaluate strategies to manage the associated anxiety and depression. Other research needs include the development and assessment of standardised care pathways and communication tools, and studies to evaluate lifestyle approaches to disease modification and impact of ADPKD centres on outcomes and costs (Chapman et al. 2015).
Conclusion
This is an important time in the ADPKD field. Several ongoing developments offer the potential for new standards of care, but national and international collaborative efforts that involve and empower patients are needed to ensure they translate into benefit for patients across Europe. To this end, the EAF aims to facilitate dialogue and collaboration
between patients and their representative organisations, nephrologists and other specialist physicians, geneticists, healthcare system managers, national government health ministries and bodies responsible for medicines regulation and HTA.
Acknowledgements
Richard Sandford and Tess Harris are co-chairs of the European ADPKD Forum (EAF), an independent, multidisciplinary, international faculty of ADPKD experts dedicated to improving the health and quality of life of people with ADPKD. For further information, please refer to the 2015 EAF Report, Translating science into policy to improve ADPKD care in Europe (EAF 2015), available at www. pkdinternational.org/EAF_ADPKD_Policy_Report_2015. The EAF was initiated by, and is solely supported by, Otsuka Pharmaceutical Europe Ltd. Neither the Co-Chairs of the EAF, nor the Faculty members, receive fees in respect of their roles in the initiative. The contents of the EAF Report are the opinions of the EAF Faculty and do not necessarily represent those of Otsuka.

References:
Ars E, Bernis C, Fraga G et al. (2014) Spanish guidelines for the management of autosomal dominant polycystic kidney disease. Nephrol Dial Transplant, 29(Suppl. 4): iv95–105.
Carr A, Makin A, Baker A et al. (2014) Do we under-estimate the physical and emotional impact of early stage ADPKD? Evidence for a discrepancy between patient experience and physician perceptions. 51st ERA-EDTA Congress, Amsterdam, Netherlands, 31 May–3 June 2014: Abstract/poster SP020. [Accessed: 5 June 2015] Available from http://ndt.oxfordjournals.org/content/29/suppl_3.toc
Chang MY, Ong AC. (2013) New treatments for autosomal dominant polycystic kidney disease. Br J Clin Pharmacol, 76(4): 524–35.
Chapman AB, Devuyst O, Eckardt KU et al. (2015) Autosomal-dominant polycystic kidney disease (ADPKD): executive summary from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int, 88(1): 17-27.
Casteleijn NF, Visser FW, Drenth JP et al. (2014) A stepwise approach for effective management of chronic pain in autosomal-dominant polycystic kidney disease. Nephrol Dial Transplant, 29(Suppl 4): iv142–53.
European ADPKD Forum (EAF) (2015) Translating science into policy to improve ADPKD care in Europe, EAF, 2015 [Accessed: 5 June 2015] Available from http://www.pkdinternational.org/eaf_adpkd_forum_policy_report_2015/
Gorriz JL, Montomoli M, Castro C et al. (2014) Adult polycystic kidney disease patients show a very similar cardiovascular comorbidity profile to CKD patients with other etiologies. Most cardiovascular and renal risk factors are not controlled in the majority of patients (Abs SA-PO558). J Am Soc Nephrol, 25(Abs Suppl): 764A.
HTA International (2015) Good practice examples of patient and public involvement in health technology
assessment. [Accessed: 5 June 2015] Available from http://www.htai.org/fileadmin/HTAi_Files/ISG/patientInvolvement/EffectiveInvolvement/Good_Practice_Examples_Feb_2015.pdf
Kerr M, Bray B, Medcalf J et al. (2012) Estimating the financial cost of chronic kidney disease to the NHS in England. Nephrol Dial Transplant, 27(Suppl 3): iii73-–80.
Oberdahn D, Palsgrove AC, Cole JC et al. (2014) Patient experience with pain related to autosomal dominant polycystic kidney disease (ADPKD) [Abs PUB285]. J Am Soc Nephrol, 25(Abs Suppl): 960A.
Ong AC, Deyust O, Knebelmann B, et al. (2015) Autosomal dominant polycystic kidney disease: the changing face of clinical management. Lancet, 385(9981): 1993–2002. Otsuka Pharmaceutical Europe Ltd. Jinarc 15 mg tables. Summary of product characteristics. 2015. [Accessed: 5 June 2015] Available from http://ec.europa.eu/health/documents/community-register/2015/20150527131681/anx_131681_en.pdf
Palmer SC, de Berardis G, Craig JC et al. (2014) Patient satisfaction with in-centre haemodialysis care: an international survey. BMJ Open, 4(5): e005020.
Petzold K, Gansevoort RT, Ong AC et al. (2014) Building a network of ADPKD reference centres across Europe: the EuroCYST initiative. Nephrol Dial Transplant 29(Suppl 4): iv26–32.
Phelps RG, Taylor J, Simpson K et al (2014) Patients’ continuing use of an online health record: a quantitative evaluation of 14,000 patient years of access data. J Med Internet Res, 16(10): e241.
Schrier RW, Abebe KZ, Perrone RD et al. (2014) Blood pressure in early autosomal dominant polycystic kidney disease. N Engl J Med, 371(24): 2255–66.
Simms RJ, Travis DL, Durkie M et al. (2015) Genetic testing in the assessment of living related kidney
donors at risk of autosomal dominant polycystic kidney disease. Transplantation, 99(5): 1023–9.
Spithoven EM, Kramer A, Meijer E et al. (2014a) Renal replacement therapy for autosomal dominant polycystic kidney disease (ADPKD) in Europe: prevalence and survival-an analysis of data from the ERAEDTA Registry. Nephrol Dial Transplant, 29(Suppl 4): iv15–iv25.
Spithoven EM, Kramer A, Meijer E et al. (2014b) Analysis of data from the ERA-EDTA Registry indicates that conventional treatments for chronic kidney disease do not reduce the need for renal replacement therapy in autosomal dominant polycystic kidney disease. Kidney Int, 86(6): 1244–52.
Tong A, Rangan GK, Ruospo M et al. (2015a) A painful inheritance – patient perspectives on living with polycystic kidney disease: thematic synthesis of qualitative research. Nephrol Dial Transplant, 30(5): 790–800.
Tong A, Chando S, Crowe S et al. (2015b) Research priority setting in kidney disease: a systematic review. Am J Kidney Dis, 65(5): 674–83.
Torres VE , Chapman AB, Devuyst O et al. (2012) Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med, 367(25): 2407–18.
Torres VE, Harris PC, Pirson Y et al. (2007) Autosomal dominant polycystic kidney disease. Lancet, 369(9569): 1287–301.
Van Biesen W, van der Veer SN, Murphey M et al. (2014) Patients’ perceptions of information and education for renal replacement therapy: an independent survey by the European Kidney Patients’ Federation on information and support on renal replacement therapy. PLoS One, 9(7): e103914.
Woywodt A, Vythelingum K, Rayner S et al. (2014) Single-centre experience with Renal PatientView, a web-based system that provides patients with access to their laboratory results. J Nephrol, 27(5): 521-7.
Youssouf S, Harris T, O’Donoghue D (2015) More than a kidney disease: a patient-centred approach to improving care in autosomal dominant polycystic kidney disease. Nephrol Dial Transplant, 30(5): 693–5.














